
Отчет Научного совета по катализу за 2004 г.
М.Г. Слинько "Эссе о нелинейности в катализе"
Терминология в гетерогенном катализе
под ред. Р.П.Бурвелла
Русский перевод
Ориганальная версия
За рубежом
Приглашения на конференции
1.1 Катализ и катализаторы
Катализ - это явление, в котором инородный материал, названный катализатором, присутствуя в относительно малом количестве и не расходуясь, увеличивает скорость химической реакции. То же относится к реагентам, для которых добавление некоторого вещества уменьшает скорость определенной реакции, например введение ингибитора в цепную реакцию или яда в каталитическую реакцию. Термин "отрицательный катализ" использовался для описания этих явлений, но теперь он не рекомендуется; предпочтительными являются такие термины, как ингибирование или отравление.
Катализатор обеспечивает протекание ряда элементарных процессов (часто называемых элементарными стадиями), которые связывают реагенты и продукты реакции и которые не протекают в отсутствие катализатора. Например, предположим, что реакция
А = С
идет с некоторой скоростью, которая может быть измеримой, но может быть близкой к нулю. Добавление Х может обеспечить новый маршрут, включающий интермедиат В,
|
А + Х |
|
|
В |
|
Если реакция идет по этому маршруту со скоростью, значительной по сравнению со скоростью некатализируемой реакции, так что суммарная скорость возрастает, то Х - катализатор. В этом смысле каталитическая реакция - это замкнутая последовательность элементарных стадий, подобная стадиям продолжения газофазной цепной реакции.
Катализатор вступает в реакцию, но регенерируется в конце каждого реакционного цикла. Таким образом, одна единица катализатора обеспечивает конверсию многих единиц реагентов (но см. §1.7).
Конечно, катализатор может ускорять только одну или некоторые из нескольких термодинамически возможных реакций.
Трудно разделить природу на отдельные составляющие и, вероятно, ни одно из рабочих определений катализа не может быть полностью удовлетворительным. Так, вода может облегчить реакцию между двумя твердыми веществами, растворяя их. Это явление может казаться примером катализа, но такой эффект растворителя, в общем случае, не рассматривается в рамках катализа. Кинетический солевой эффект в растворе также обычно исключается из рассмотрения. Далее, катализатор должен быть веществом и, несмотря на то, что подвод теплоты к системе обычно увеличивает скорость реакции, теплоту не называют катализатором, также как и свет, вызывающий реакцию между хлором и водородом, не является катализатором.
Следует различать катализатор и инициатор. Инициатор начинает цепную реакцию, например, перекись ди-т-бутила в полимеризации стирола, но инициатор расходуется в реакции. Это не катализатор.
В гомогенном катализе все реагенты и катализатор молекулярно диспергированы в одной фазе.
В гетерогенном катализе катализатор образует отдельную фазу. Обычно катализатор является кристаллическим или аморфным твердым веществом, тогда как реагенты и продукты находятся в одной или более жидких фазах. Каталитическая реакция идет на поверхности твердого вещества и, в идеале, ее скорость пропорциональна поверхности катализатора. Однако на практике скорость может ограничиваться транспортными процессами (см. §1.6).
Большинство примеров катализа легко различаются как гомогенные и гетерогенные, но некоторые находятся на пересечении этих двух типов. Рассмотрим систему, в которой интермедиаты образуются на поверхности, а затем десорбируются в газовую фазу. Такие интермедиаты могут генерировать цепную реакцию в газовой фазе, т.е. инициация и обрыв цепи происходят на поверхности, но рост цепи - в газовой фазе.
Катализ ферментами может обладать некоторыми характеристиками как гомогенного, так и гетерогенного катализа, когда катализатором является макромолекула, которая достаточно мала, чтобы быть молекулярно диспергированной в одной фазе со всеми реагентами, но достаточно велика, чтобы говорить об активных центрах на ее поверхности.
Данное руководство посвящено гетерогенному катализу. Другие типы катализа далее не будут рассматриваться.
1.2 Адсорбция
1.2.1 Общие термины
Несмотря на то, что адсорбция существует как объект научных исследований, независимый от ее роли в гетерогенном катализе, она требует особого внимания в рамках данного руководства, благодаря ее определяющей роли в гетерогенном катализе. Большинство или почти все каталитические реакции включают адсорбцию по крайней мере одного из реагентов. Многим терминам, относящимся к адсорбции, уже были даны определения в Приложении II, Части I, §1.1. (Справочник символов и терминологии по физико-химическим величинам и единицам, подготовленный ранее комиссией по коллоидной и поверхностной химии, IUPAC) Они включают поверхность, поверхность раздела, площадь поверхности или поверхности раздела и удельную площадь поверхности. Приложение II, Часть I рекомендует A или S и a или s как обозначения, соответственно, площади поверхности и удельной площади поверхности. As и as могут использоваться там, где это необходимо, во избежание путаницы с энергией Гельмгольца A или энтропией S.
Другими терминами являются сорбция, сорбтив и сорбат [различая вещество в сорбированном состоянии (сорбат) и вещество в текучей фазе, которое может быть сорбировано (сорбтив)], абсорбция, абсорбтив, абсорбат, абсорбент; и адсорбция, адсорбтив, адсорбат, адсорбент(Использование термина субстрат для обозначения адсорбента или носителя должно обескураживать, так как обычно используется в химии ферментов для обозначения реагентов) . Термин адсорбированный комплекс используется для обозначения объекта, образованного адсорбатом и той частью адсорбента, с которой он связан.
Приложение II, Часть I, §1.1.5 следующим образом рассматривает поверхность раздела адсорбент/флуид (подвижная среда) (ПРиложение II, Часть I рекомендует. Косая черта предпочтительнее, чем дефис, для разделения названий объемных фраз, т.к. использование дефиса может привести к двусмысленности) . "Зачастую полезно рассматривать поверхность раздела адсорбент/флуид как содержащую две области. Область флуидной (текучей) фазы (т.е. жидкость или газ) как часть этой поверхности раздела может быть названа областью адсорбции, тогда как часть адсорбента, включенная в поверхность раздела, называется поверхностным слоем адсорбента".
Для процесса, в котором молекулы* или диссоциированные молекулы накапливаются в адсорбционном пространстве или в приповерхностном слое абсорбента, термином, противопоставленным адсорбции, будет десорбция, обозначающая обратный процесс (см. Приложение II, Часть I, §1.1.4). Адсорбция также используется для обозначения результата процесса адсорбции, т.е. присутствия адсорбата на адсорбенте. Адсорбированное состояние может быть или не быть в равновесии с адсорбтивом [см. 1.2.2(с)].
Адсорбция и десорбция также указывают направление достижения равновесия, например, кривая (точка) адсорбции, кривая (точка) десорбции.
1.2.2 Хемосорбция и физосорбция
Соответствующие фрагменты §§1.1.6 и 1.1.7 Приложения II, Части I приведены ниже.
"Хемосорбция и физосорбция"
Хемосорбция (или химическая адсорбция) - это адсорбция, в которой действуют такие же валентные силы, как и при образовании химических соединений. Разграничение хемосорбции и физосорбции является, по сути, проблемой, сходной с разграничением химического и физического взаимодействия вообще. Абсолютно четкое различие невозможно; существуют промежуточные случаи, например, адсорбция, включающая сильные водородные связи или слабый перенос заряда.
Отличить хемосорбцию можно по некоторым признакам, в том числе следующим:
Физосорбция (или физическая адсорбция) - это адсорбция, при которой действуют те же межмолекулярные силы (силы Ван дер Ваальса), что и силы, отвечающие за неидеальность реальных газов и конденсацию паров, и которые не вызывают существенные изменения электронных орбиталей адсорбированных молекул. Термин Ван дер Ваальсова адсорбция, хотя и является синонимом термина физическая адсорбции, для использования не рекомендуется.
Распознать физическую адсорбцию можно по некоторым признакам, в том числе:
Монослойная и многослойная адсорбция, заполнение микропор и капиллярная конденсация
При монослойной адсорбции все адсорбированные молекулы находятся в контакте с поверхностным слоем адсорбента.
При многослойной адсорбции адсорбционное пространство содержит более одного слоя молекул, и не все адсорбированные молекулы находятся в контакте с поверхностным слоем адсорбента.
Монослойной емкостью называют в случае хемосорбции количество адсорбата, необходимое для заполнения всех адсорбционных центров, определяемых структурой адсорбента и химической природой адсорбтива, а в случае физосорбции - количество адсорбата,
необходимое для покрытия поверхности заполненным монослоем молекул в плотной упаковке, причем плотная упаковка при необходимости должна быть явно выражена. Величины, относящиеся к монослойной емкости, могут обозначаться подстрочным индексом m.
Покрытие поверхности(q ), в случае как монослойной, так и многослойной адсорбции, определяется как отношение количества адсорбированного вещества к монослойной емкости.
Площадь, занимаемая одной молекулой в заполненном монослое, обозначается am; например, для молекул азота am (N2).
Заполнение микропор - это процесс, в котором молекулы адсорбируются в адсорбционном пространстве внутри микропор.
Объем микропор обычно измеряется по объему адсорбированного материала, заполняющего микропоры полностью, и выражается в единицах объема жидкости при атмосферном давлении и температуре измерений.
В определенных случаях (например, пористые кристаллы) объем микропор может быть найден из структурных данных.
Говорят, что в пористых твердых телах имеет место капиллярная конденсация, когда полимолекулярная адсорбция из газовой фазы происходит до точки, в которой пористое пространство заполнено жидкостью, отделенной менисками от газовой фазы.
Понятие капиллярной конденсации теряет смысл, когда размеры пор настолько малы, что термин "мениск" уже не имеет физического смысла. Капиллярная конденсация часто сопровождается гистерезисом."
1.2.3 Типы хемосорбции
Недиссоциативная и диссоциативная хемосорбция. Если молекула адсорбируется без фрагментирования, то адсорбционный процесс является недиссоциативным. К такому типу часто относится адсорбция моноокиси углерода. Если адсорбируемая молекула диссоциирует на два или более фрагментов, оба или все из которых связаны с поверхностью адсорбента, такой процесс называется диссоциативным. Хемосорбция водорода относится преимущественно к такому типу.
H2(g) 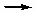 2H (ads) или H2(g) + 2* 2H*
2H (ads) или H2(g) + 2* 2H*
Звездочка обозначает поверхностный центр.
Термины гомолитический и гетеролитический относятся обычно к формальной природе разрыва одинарной связи. Если в процессе диссоциативной адсорбции электронная пара химической связи адсорбтива А : В разделяется, то это гомолитическася диссоциативная адсорбция. Если же электронная пара остается у А или В, то говорят о гетеролитической диссоциативной адсорбции.Ниже даны примеры.
(а) Гомолитическася диссоциативная адсорбция водорода на поверхности металла:
H2 + 2* 2H*.
(b) Гетеролитическая диссоциативная адсорбция водорода на поверхности оксида, в котором для поверхностных центров Mn+ и O2 - характерна более низкая координация, чем для ионов в объеме фазы:
H2 + Mn+ + O2 - H - Mn+ + HO - .
При необходимости внесения еще большей определенности это уравнение может быть записано как
H2(g) + Msn+ + Os2 - H - Msn+ + HOS - ,
где подстрочный индекс s указывает на то, что данные группы принадлежат поверхности.
Обозначение H - Mn+ используется, как и в традиционной терминологии неорганической химии, для указания сохранения степени окисления М.
(c) Гетеролитическая диссоциативная адсорбция воды на такой же паре центров, как и в примере (б):
H2O + Mn+ + O2 - HO - Mn+ + HO - .
Восстановительная и окислительная диссоциативная адсорбция аналогичны терминам, используемым в координационной химии, которые, например, описывают следующую реакцию как окислительное присоединение
L4M(I) + H2 L4M(III)H2.
Здесь М - атом переходного металла, а L - лиганд. Н как лиганд дан в степени окисления - 1. При восстановительном присоединении электронная пара, образующая связь в сорбтиве А : В, переносится к поверхностным соединениям; в случае окислительного присоединения электронная пара удаляется из поверхностных соединений. Можно сказать, что диссоциативная адсорбция Cl2 на металле является окислительной, если на поверхности адсорбента хлор образует ионы Cl - . Диссоциативная адсорбция будет восстановительной, если, например, идет таким образом:
H2(g) + 2[M(III)O2 - ]s 2[M(II)(OH) - ]s.
Заметим, что здесь H2 2H+ + 2e.
Адсорбция с переносом заряда - это разновидность окислительной или восстановительной хемосорбции, в которой слова "восстановительная" или "окислительная" относятся к приобретению или потере электрона группами твердого тела. В простых случаях она недиссоциативна, т.е. происходит просто перенос заряда между адсорбривом и адсорбентом с образованием адсорбата. Ниже даны два примера.
pВосстановительная: X + * X+* -
где Х - ароматическая молекула с низким потенциалом ионизации, такая как антрацен или трифениламин, а * - центр на алюмосиликате.
Окислительная: О2 + * О2 - * +
Термин "адсорбция с переносом заряда" применялся также к адсорбции, напоминающей комплексы Малликена с переносом заряда.
Иммобильный, мобильный. Эти термины используются для описания свободы перемещения молекул адсорбата по поверхности. Адсорбция называется иммобильной, когда значение kT меньше величины D E энергетического барьера, разделяющего близлежащие центры. В этом случае у адсорбата мало возможности мигрировать по соседним центрам, и такая адсорбция обязательно локализованная. Подвижность адсорбата будет возрастать с повышением температуры, и мобильная адсорбция может быть как локализованной, так и нелокализованной. В случае локализованной мобильной адсорбции адсорбат основное время находится на адсорбционных центрах, но может мигрировать или десорбироваться с последующей реадсорбцией на других местах. При нелокализованной адсорбции подвижность настолько велика, что лишь малая часть адсорбированных групп находится на адсорбционных центрах, в то время как большая часть находится на других местах поверхности.
В некоторых случаях локализованной адсорбции адсорбат упорядочен в двумерную решетку или сеть в некоторой области значений покрытия поверхности и температур. Если сеть упорядоченной адсорбированной фазы соответствует решетке адсорбента, то структуру называют когерентной, в противном случае это некогерентная структура (см. также §2.4).
Каждому из типов процессов адсорбции может соответствовать десорбция в обратной форме, например процесс, обратный диссоциативной адсорбции - это ассоциативная десорбция. Тем не менее, процесс хемосорбции может быть и необратимым [§1.2.2(с)]. В результате десорбции могут образовываться соединения, отличные от адсорбированных, например, при десорбции этана, диссоциативно адсорбированном на чистом никеле, очень мало или вообще не образуется этан; 1-бутен, диссоциативно адсорбированный на окиси цинка с образованием метилаллила и Н, дает при десорбции 2-бутены, а с поверхности вольфрама, покрытой адсорбированным кислородом, может испаряться WO3.
Фотоадсорбция, фотодесорбция. Световое излучение (обычно в видимой или ультрафиолетовой областях) может воздействовать на адсорбцию. В системе, содержащей адсорбтив и адсорбент, воздействие света может приводить к увеличению адсорбции (фотоадсорбция) или к десорбции адсорбата (фотодесорбция).
1.2.4 Центры хемосорбции
Классификация центров может производиться исходя из их химической природы в соответствии с обычной химической терминологией. Следующие термины являются простым расширением области обычного использования химических терминов: оснoвные центры ,кислотные центры льюисовские кислотные центры,протонные или бренстедовские кислотные центры,электроноакцепторные центры и электроннодонорные центры (два последних примера могут возникать в связи с адсорбцией с переносом заряда).
Нередко полезно учитывать, что центры хемосорбции являются результатом координационной ненасыщенности поверхности, т.е. того, что координационное число поверхностных атомов меньше координационного числа таких же атомов в объеме. Так, например, ион хрома на поверхности окиси хрома имеет координационное число меньшее, чем ион хрома в объеме. Поэтому этот ион хрома будет стремиться к образованию связи с подходящим адсорбтивом, чтобы восстановить свое координационное число. Координационное число атома, находящегося на плоскости (100) гранецентрированного кубического металла, равно 8 по сравнению с числом 12 для атома в объеме металла; это тоже является примером координационной ненасыщенности поверхности. Однако, конечно, существуют центры, к которым понятие поверхностной координационной ненасыщенности неприменимо, например, это бренстедовские кислотные центры.
Зачастую нельзя с уверенностью идентифицировать тип и структуру центров в адсорбции и гетерогенном катализе. Однако для теоретического рассмотрения возможных центров необходимо введение ряда обозначений. С одной стороны, хотелось бы иметь возможность использовать общее и неспецифическое описание. Для этого можно порекомендовать символы * и (ads), например, H* и H(ads). С другой стороны, может возникнуть желание использовать как можно более определенные обозначения. В этом случае могут оказаться полезными общехимические символы. Набор символов для металлов включает Cj и Bn, где Cj обозначает поверхностный атом, имеющий j ближайших соседей, а Bn - ансамбль из n поверхностных атомов, которые в совокупности образуют адсорбционный центр, например, адсорбционный центр между тремя поверхностными атомами, образующими углы равностороннего треугольника, является центром типа B3 [более подробно см. van Hardeveld and Hartog, Surface Sci. 15, 189(1969)].
Известны случаи хемосорбции, когда при высокой степени покрытия сеть (двумерная решетка) адсорбата не совпадает с решеткой адсорбента. В таком случае понятие центров с точной локализацией и их фиксированным количеством может оказаться неприменимым. Схожие проблемы в определении центров возникают и в случае перестройки поверхности при взаимодействии адсорбата с адсорбентом. Из-за трудностей, нередко возникающих при определении типа поверхностных центров, часто становится удобным, особенно для металлов, определять покрытие поверхности q как отношение числа адсорбированных атомов или групп к числу поверхностных атомов (сравни §1.2.2).
1.2.5 Однородность центров
Природа центров адсорбции или катализа может меняться даже в случае чистых металлов, где не возникает вопроса относительно различий химического состава той или другой части поверхности. Такие изменения возникают не только вследствие дефектов на поверхности металла, но и потому, что природа центра зависит от структуры поверхности. Однородные центры скорее всего встречаются, когда адсорбция или катализ изучаются на индивидуальной грани монокристалла; тем не менее даже индивидуальные грани могут содержать более одной разновидности центров. Неоднородные центры будут наблюдаться, в качестве нормального явления, в образцах металлов с более чем одним типом доступных кристаллических граней. Различают два вида неоднородности. Собственная неоднородность обусловлена только природой адсорбента. Наведенная(индуцированная) неоднородность возникает в том случае, когда присутствие молекулы адсорбата на одном из центров приводит к изменению силы адсорбции на соседних центрах. Таким образом, совокупность однородных центров на индивидуальной кристаллической грани может стать неоднородной в случае частичного заполнения поверхности хемосорбированными частицами.
При исследовании каталитических свойств металлов важность неоднородности центров зависит от особенностей изучаемой реакции. Для некоторых реакций активность металлического катализатора зависит только от суммарного количества доступных центров; эти реакции называются структурно-нечувствительными. В других реакциях, относящихся к структурно-чувствительным, активность может быть намного выше на центрах, связанных с определенной кристаллической гранью или даже с некоторыми типами структурных дефектов. Для обозначения структурно-нечувствительных и структурно-чувствительных реакций использовались также альтернативные термины легкие и затрудненные реакции, соответственно. Термины в §1.2.5 обсуждались в отношении к поверхности металлов, тем не менее их можно использовать и по отношению к другим адсорбентам и катализаторам, особенно к парным центрам, участвующим в гетеролитической диссоциативной адсорбции.
1.2.6 Активные места, активные центры (Active site, active centre
Термин активные места (active sites) зачастую применяется к центрам адсорбции, являющимся эффективными местами определенной гетерогенной каталитической реакции. Термины активные места и активные центры(active site и active centre) часто используются как синонимы, но термин активный центр может применяться также и для описания ансамбля мест (sites), на которых протекает каталитическая реакция.
1.2.7 Изотермы адсорбции
Изотерма адсорбции простого газообразного адсорбтива на твердом теле является функцией, связывающей при постоянной температуре количество вещества, адсорбированного в равновесии с давлением (или концентрацией) адсорбтива в газовой фазе. Величиной, доступной для экспериментальных измерений, является скорее поверхностный избыток, а не адсорбированное количество, тем не менее при низких давлениях разница между этими двумя величинами становится пренебрежимо малой (см. Приложение II, Часть I, §1.1.11).
Аналогичным образом, когда два или более адсорбтива конкурируют за адсорбцию на поверхности, изотерма адсорбции адсорбтива i при данной температуре есть функция равновесных парциальных давлений адсорбтивов всех типов. В случае адсорбции из жидкого раствора изотерма адсорбции любого преимущественно адсорбированного вещества из раствора может быть определена сходным образом c помощью равновесной концентрации соответствующего компонента раствора, однако изотерма обычно зависит от природы растворителя и от концентраций (мольных долей) других растворенных компонентов, если они есть. Индивидуальные изотермы растворенных веществ не могут быть выведены из поверхностных избытков иначе как с использованием соответствующей модели адсорбированного слоя; когда имеет место хемосорбция, в общем случае можно предположить, что адсорбция монослойная. Количества адсорбированного вещества часто выражаются через степень покрытия q i. В случае хемосорбции q i - это доля центров адсорбции, покрытых веществом i. Ниже описаны типы изотерм адсорбции, представляющие интерес для гетерогенного катализа. Линейная изотерма адсорбции. Простейшая изотерма адсорбции является аналогом закона Генри. В случае единственного адсорбтива она принимает вид
q = Kp или q = Kc,
где p и c - давление и концентрация адсорбтива, q - покрытие адсорбатом, а K - константа равновесия линейной изотермы адсорбции или константа Генри. Большинство изотерм адсорбции сводятся к закону Генри, когда p или c становятся достаточно малыми при условии, что имеет место простая адсорбция, т.е. адсорбция не является ни диссоциативной, ни ассоциативной. То есть при достаточно малом покрытии закон Генри обычно применяется лишь к первому из последующих уравнений, но не ко второму или третьему.
A + * 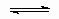 A*;
A*;
A2 + 2* 2A*;
2A + * A2*.
Изотерма адсорбции Лэнгмюра,
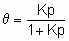 or
or 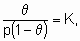
или ее эквиваленты в терминах концентраций обычно имеют место при простой (недиссоциативной) адсорбции из идеального газа на поверхности с фиксированным числом однородных центров, которые могут быть заняты одним, и только одним видом адсорбата. K называется константой равновесия адсорбции по Лэнгмюру. Кроме того, энтальпия адсорбированной формы не должна зависеть от того, заняты или нет соседние центры адсорбции, и, следовательно, энтальпия адсорбции не зависит от q
. Вторая из приведенных выше форм изотермы Лэнгмюра подчеркивает, что константа K является константой равновесия реакции A + * A*. Поскольку постоянство энтальпии при любом покрытии поверхности аналогично постоянству энтальпии при изменении давления идеального газа, адсорбционное состояние в системе, подчиняющейся изотерме адсорбции Лэнгмюра, иногда называют идеальным адсорбционным (адсорбированным) состоянием В случае диссоциативной хемосорбции,
A2 + 2* 2A*
уравнение Лэнгмюра принимает вид
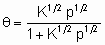 или
или 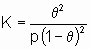
Для простой адсорбции двух адсорбтивов A и B, конкурирующих за одни и те же центры, изотерма Лэнгмюра принимает вид
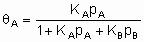 ,
,
где KA и KB - константы равновесия раздельной адсорбции А и В, соответственно. Это уравнение может быть обобщено на случай описания адсорбции нескольких адсорбтивов и может учитывать диссоциативную адсорбцию одного и более адсорбтивов.
В изотерме адсорбции Фрейндлиха количество адсорбированного вещества пропорционально дробной степени давления адсорбтива. В некоторых системах дробная степень и константа пропорциональности являются функциями температуры. С учетом покрытия изотерма принимает вид
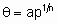 ,
,
где n - число, большее единицы, и a - константа. В области справедливости изотермы (дифференциальная) энтальпия адсорбции является линейной функцией от ln q .
В изотерме адсорбции Темкина количество адсорбированного вещества соотносят с логарифмом давления адсорбтива
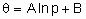 ,
,
где A и B константы. В области справедливости такой изотермы (дифференциальная) энтальпия адсорбции является линейной функцией q . Изотерма адсорбции Брунауера-Эммета-Теллера применима только к физосорбции паров, но важна также и для гетерогенного катализа, поскольку используется для определения поверхности твердых тел. Изотерма описывается следующим уравнением
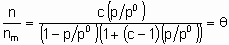 ,
,
где с - константа, зависящая от температуры и природы адсорбтива и адсорбента, n - количество адсорбированного вещества, nm - емкость монослоя и p0 - давление насыщенных паров жидкого адсорбтива при температуре исследования. В соответствии с этим уравнением, основанным на модели многослойной адсорбции, q
превышает единицу, если отношение 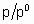 достаточно велико.
достаточно велико.
1.2.8 Бифункциональный катализ
Некоторые гетерогенные каталитические реакции протекают как последовательность элементарных процессов, определенная часть которых локализуется на центрах одного типа, тогда как другие - на центрах совершенно другой природы. Например, некоторые из реакций процесса риформинга углеводородов на катализаторах платина/оксид алюминия протекают на поверхности платины, а другие - на кислотных центрах оксида алюминия. О таких каталитических реакциях говорят как о бифункциональном катализе. Оба типа центров обычно перемешаны в одной и той же первичной частице (§1.3.2), однако те же реакции могут идти даже при использовании смеси частиц, каждая их которых содержит только один тип центров. Такой подход, конечно, может быть расширен, чтобы создать понятие полифункционального катализа.
1.2.9 Скорости адсорбции и десорбции
Коэффициент прилипания - это отношение скорости адсорбции к скорости соударений адсорбтива со всей поверхностью, т.е. как заполненной, так и не заполненной. Обычно это функция покрытия поверхности, температуры и деталей структуры поверхности адсорбента. Вероятность прилипаниячасто используется в таком же смысле, но, в принципе, это микроскопическая величина, относящаяся к процессу индивидуального столкновения. Таким образом, коэффициент прилипания можно рассматривать как среднюю вероятность прилипания, усредненную как по всем углам и энергиям ударяющихся молекул, так и по всей поверхности.
Среднее время задержки (пребывания) адсорбированных молекул - это среднее время, в течение которого молекула остается на поверхности адсорбента, т.е. средний интервал времени между ударом и десорбцией. В то время, когда молекула остается на поверхности, до десорбции она может мигрировать между адсорбционными центрами по поверхности. Если время задержки адсорбированных групп относится к определенным адсорбционным центрам, его можно называть средним временем жизни определенного адсорбционного комплекса. Когда скорость десорбции имеет первый порядок по покрытию, время пребывания не зависит от покрытия поверхности и обратно пропорционально константе скорости процесса десорбции. В этом случае оно также может быть однозначно определено с помощью времени полураспада (полужизни) или какого-либо другого дробного времени жизни десорбционных процессов. Если процесс десорбции не является процессом первого порядка, например, из-за взаимовлияния адсорбированных молекул и/или энергетической неоднородности поверхности, время пребывания зависит от покрытия поверхности, и рабочая формулировка "времени пребывания" требует более конкретного определения.
Неактивированная и активированная адсорбция. Если температурный коэффициент скорости адсорбции очень мал, говорят о неактивированном процессе адсорбции (т.е. имеющем пренебрежимо малую энергию активации). В этом случае коэффициент прилипания при низком покрытии может быть близок к единице, особенно для молекул меньшего размера. Если температурный коэффициент скорости адсорбции имеет существенную величину, то говорят, что процесс - активированный (т.е. имеющий значительную энергию активации). В этом случае коэффициент прилипания мал. В общем случае, энергия активации активированной адсорбции является функцией покрытия и, как правило, возрастает с увеличением покрытия.
Предположены различные соотношения между скоростью активированной адсорбции и покрытием. Особенно часто используют одно из них, а именно уравнение Рогинского-Зельдовича, иногда называемое также уравнением Еловича,
где q - это покрытие, а a и b - константы, характерные для конкретной системы.


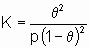
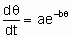 ,
,