
Терминология в гетерогенном катализе
под ред. Р.П.Бурвелла (продолжение)
Русский перевод
Ориганальная версия
О шестой рамочной программе Евросоюза
За рубежом
Приглашения на конференции
(Продолжение, начало см. "Каталитический Бюллетень" N1(33), 2005, с. 24-61)
1.3 Состав, структура и текстура катализаторов
1.3.1 Общие термины
Катализаторы могут быть однофазными или двухфазными. В первом случае они могут содержать одно вещество (например, оксид алюминия или платиновую чернь) или быть однофазным раствором одного или более веществ. В этом случае компоненты раствора должны представляться соединенными дефисом (например, окись кремния-окись алюминия).
Носитель. В многофазных катализаторах каталитически активный материал часто присутствует в малом количестве как компонент, диспергированный на носителе (support или, иногда, carrier). Носитель может быть каталитически инертным, но вносить вклад в суммарную каталитическую активность. Предельный случай - это некоторые бифункциональные катализаторы (§1.2.8). Такой катализатор следует называть, начиная с активного компонента, на втором месте следует носитель, и эти два слова или фразы разделяются косой чертой, например, платина/окись кремния или платина/алюмосиликат. Косая черта иногда заменяется словом "на", например, платина на окиси алюминия.
Промотор. В некоторых случаях добавление относительно небольшого количества одного или более веществ, промотора или промоторов, к катализатору повышает активность, селективность или полезное время работы катализатора. В общем случае промотор может либо ускорить целевую реакцию, или подавить нежелательную. Не существует формальной номенклатурной системы для обозначения промотированных катализаторов. Можно, однако, использовать фразу "железо, промотированное окисью алюминия и окисью калия".
Промотор, действие которого направлено на уменьшение тенденции к спеканию и уменьшению поверхности, может быть назван текстурным промотором (см. §1.7.3).
Легирование. В случае полупроводящих катализаторов, небольшое количество примесного материала, растворенного в исходном катализаторе, может изменять скорость определенной реакции. Это явление иногда называется легированием по аналогии с действием подобного материала на полупроводимость.
.1.3.2 Пористость и текстура
Многие, но не все катализаторы - пористые материалы, в которых большая часть площади поверхности является внутренней. Иногда удобно говорить о структуре и текстуре таких материалах. Структура определяется как распределение в пространстве атомов или ионов в материальной части катализатора, и особенно как распределение по поверхности. Текстура определяется как детальная геометрия пустого пространства в частицах катализатора. Пористость - это понятие, связанное с текстурой и относящееся к пористому пространству в материале. Однако в случае цеолитов пористость, в основном, определяется кристаллической структурой.
Точное описание текстуры пористого катализатора потребовало бы детального определения очень большого числа параметров. Часто используются следующие усредненные свойства.
В случае пористых твердых веществ поверхность, связанная с порами, может быть названа внутренней поверхностью. Так как доступность пор может зависеть от размера молекул жидкости или газа, протяженность доступной внутренней поверхности может зависеть от размера молекул, составляющих жидкость или газ, и может различаться для компонентов жидкой или газообразной смеси (эффект молекулярного сита).
Когда пористое тело состоит из дискретных частиц, удобно описывать наружную границу частиц как внешнюю поверхность.
Целесообразно классифицировать поры в соответствии с их размерами:
Не рекомендуются термины промежуточные или переходные поры, которые часто использовались в прошлом.
В случае микропор их суммарный доступный объем может рассматриваться как адсорбционное пространство.
Вышеперечисленные ограничения в некоторой степени произвольны. В определенных обстоятельствах может оказаться удобным выбрать какие-либо другие величины.
Распределение пор по размерам - это распределение объема пор по размерам пор; другими словами оно может быть определено как соответствующее распределение площади по размерам пор. Это важный фактор кинетического поведения пористого катализатора и, таким образом, существенное свойство для их охарактеризования (см. §1.6).
Расчет такого распределения включает произвольные допущения, и распределение пор по размерам всегда должно сопровождаться указанием на то, какой метод использован для их определения. Обычно используются либо один, либо оба следующих метода: (i) изотермы адсорбции-десорбции азота или других адсорбтивов в сочетании с соответствующей моделью конвертирования изотермы в распределение пор по размерам; (ii) данные, полученные с помощью ртутной порометрии. Такие изотермы дают распределение для размеров мезопор. Ртутная порометрия дает распределение для макропор и более крупных мезопор. В обоих случаях то, что измерено, является, строго говоря, не точным объемом пор данного размера, а объемом пор, доступных через поры данного размера. Соотношение этих двух функций зависит от геометрической природы системы пор.
Удельный объем пор - это суммарный объем внутренних пустот на единицу массы адсорбента. Часть объема пор может быть полностью закрыта и поэтому недоступна для молекул, участвующих в каталитической реакции.
Суммарный доступный объем пор может быть измерен количеством адсорбата при давлении насыщения адсорбтива, рассчитанного как объем жидкости, при условии, что адсорбция на внешней поверхности пренебрежимо мала или может быть оценена. Доступный объем пор может различаться для молекул разного размера. Методом, на который не влияет внешняя поверхность, является определение мертвого пространства с помощью несорбируемого газа (обычно гелия) в сочетании с определением общего объема адсорбента с использованием несмачивающей жидкости или геометрических измерений.
Первичные частицы. Некоторые материалы, широко используемые как катализаторы или носители, состоят из сфероидов диаметром около 10 нм (100 Å), прочно связанных в гранулы или таблетки. По текстуре они подобны сцементированному рыхлому слою гравия. Эти частицы диаметром 10 нм (100 Å) можно назвать первичными частицами.
Степень использования (percentage exposed) металлических катализаторов. Доступность атомов металла в нанесенных или ненанесенных металлических катализаторах зависит от доли атомов от общего числа атомов металла, которые являются поверхностными. Рекомендуется это количество обозначать термином степень использования, а не часто используемым термином дисперсность.
Предварительная обработка и активация. После приготовления или загрузки в каталитический реактор катализатор часто подвергается различным обработкам перед началом каталитического процесса. Термин предварительная обработка может, в общем, применяться для набора таких обработок. В некоторых случаях используется слово активация. Оно означает, что материал превращается в катализатор или намного более эффективный материал в результате предварительной обработки. Одной из форм такой обработки является дегазация, при которой катализатор нагревается в вакууме для удаления адсорбированного или растворенного газа. Прокалка - это термин, обозначающий нагревание в воздухе или кислороде, относящийся скорее всего к стадии приготовления катализатора.
1.4 Каталитические реакторы
Сосуд, в котором проводится каталитическая реакция, называется реактором. Множество разнообразных устройств может применяться для введения реагентов и вывода продуктов.
В реакторе периодического действия реагенты и катализатор помещаются в реактор, который затем перекрывается для транспорта вещества, и реакцию проводят в течение заданного времени, после чего выводится смесь непрореагировавшего вещества и продуктов.
В проточном реакторе реагенты пропускаются через реактор, пока работает катализатор. Возможно множество вариантов.
Катализатор может быть организован в видеслоя, а реагенты пропускаются над ним. Проточный реактор со слоем катализатора обычно называется реактором с закрепленным слоем, а термин реактор идеального вытесненияиспользуется также и для указания, что не обеспечивается возвратное смешивание реакционной смеси по мере того, как она проходит через реактор. Проточный реактор работает, в основном, в дифференциальном режиме с участием небольших порций, так что состав смеси практически постоянен по длине каталитического слоя, или интегральном с участием бόльших порций, так что состав материала на выходе из слоя катализатора отличается от состава на входе.
В импульсном реакторе газ-носитель, в качестве которого может использоваться инертный газ или один из реагентов, пропускается над катализатором, а небольшие порции другого реагента или реагентов вводятся в газ-носитель через определенные промежутки времени. Импульсный реактор полезен в исследовательских работах, но кинетические результаты применимы скорее к промежуточным, чем к стационарным условиям катализатора.
Можно использовать другие режимы работы во избежание осложнений, связанных с изменением концентраций по длине каталитического слоя, связанных с интегральными проточными реакторами, каждый из режимов обозначается специальным термином. В проточном реакторе прямым перемешиванием эффективное смешивание часто обеспечивается использованием быстро вращающихся корзин, в которые загружается катализатор. Если такое перемешивание результативно, состав смеси в реакторе будет близок к составу газов на выходе.
Тот же результат может обеспечиваться рециркуляцией газа по контуру, содержащему закрепленный слой катализатора, при условии, что скорость рециркуляции намного больше скорости потока в контур и из него. В таких условиях может быть получена существенная конверсия в продукты, даже если условия в слое похожи скорее на условия дифференциального, чем интегрального реактора. Другой рабочий режим связан с использованием псевдоожиженного слоя, в котором тонкие частицы катализатора ведут себя подобно жидкости под воздействием достаточно интенсивного потока газов. Температура сохраняется постоянной во всем псевдоожиженном слое, несмотря на то, что газ и жидкость обычно не полностью смешиваются. В частности, он применяется в тех случаях, когда катализатор должен регенерироваться, например, окислением, после непродолжительного использования. Постоянный перенос катализатора между двумя сосудами (один из которых используется как реактор, а другой - для регенерации катализатора) возможно осуществить с использованием ожиженной системы. Проточные и рециркуляционные реакторы с перемешиванием характеризуются, в идеале, очень маленькими градиентами концентрации и температуры в каталитической зоне. В обоих случаях можно использовать термин безградиентный реактор.
Все реакторы, периодические или проточные, могут работать в трех основных температурных режимах. Это изотермический, адиабатическийи температурно-программируемый режимы. В последнем случае можно программировать изменения температуры во времени в реакторе периодического действия или задавать изменения температуры по длине слоя в реакторе с закрепленным слоем.
При изотермическом режиме реактор периодического действия характеризуется концентрацией адсорбатов и другими параметрами состояния поверхности, постоянными в пространстве (т.е. однородными в массе катализатора), но изменяющимися во времени. В проточном интегральном реакторе с катализатором со стационарной активностью условия на поверхности постоянны во времени, но изменяются по слою. В безградиентном реакторе в стационарном состоянии поверхностные условия постоянны в пространстве и, если катализатор находится в стационарном состоянии, во времени. В импульсном реакторе катализатор часто находится не в стационарном состоянии, концентрации изменяются по мере того, как импульс проходит через слой, а реагенты и продукты могут разделяться хроматографическими методами.
В общем случае, если гетерогенную реакцию надо проводить в изотермических условиях, конструкция реактора должна обеспечивать поток тепла к частицам катализатора или от них для того, чтобы удерживать небольшой температурный градиент. Иначе температура в слое катализатора не будет однородной. В этом смысле имеют преимущество дифференциальные реакторы и разные типы безградиентных реакторов.
Вышеописанные типы реакторов могут, в принципе, быть распространены на реакции в жидкой фазе, хотя импульсные реакторы в этом случае используются редко.
Реакции, в которых один реагент находится в газовой, а другие - в жидкой фазах с катализатором, распределенным в жидкой фазе, составляют особый, но не необычный случай, например, гидрирование жидких алкенов на платиновом катализаторе. Для лабораторных исследований таких реакций чаще всего используют реактор периодического действия. Массоперенос из газовой в жидкую фазу может снизить скорость такой каталитической реакции, если не обеспечить хороший контакт между газом и жидкостью (см. §1.6).
1.5 Кинетика гетерогенных каталитических реакций
1.5.1 Общие термины
Рассмотрим химическую реакцию
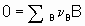
где n B - стехиометрический коэффициент (плюс для продуктов, минус для реагентов) любого продукта или реагента B. Степень реакции x определяется (см. §11.1 Руководства) как
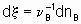
где nB - количество вещества B.
Если однозначно определить скорость реакции, то это - скорость прироста степени реакции
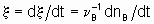
тогда как количество dnB/dt можно назвать скоростью образования(или расхода) B.
Чтобы облегчить сравнение результатов разных исследователей, скорости гетерогенных каталитических реакций должны быть должным образом выражены, а условия, при которых они были измерены, должны быть подробно описаны. Если скорость некаталитизируемой реакции пренебрежимо мала, то скорость катализируемой реакции можно определить как
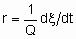
Если Q, количество катализатора - это масса,
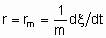
то rm - удельная скорость реакции, которую можно назвать удельной активностью катализатора в определенных условиях. Если Q - это объем,
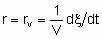
За объем принимается объем гранул без объема пространства между гранулами. Если Q - это поверхность, то
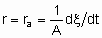
где ra - поверхостная скорость реакции. Если используется общая площадь поверхности катализатора, то предпочтительно использовать поверхность, определенную методом БЭТ с помощью адсорбции азота. Однако могут применяться и другие способы определения поверхности, например, доступная поверхность металла в нанесенных металлических катализаторах. Доступная поверхность металла часто оценивается с помощью селективной хемосорбции подходящего сорбтива, например, водорода или монокиси углерода.
Частота оборотов N,(обычно ее называют числом оборотов) определяемая, как и в катализе ферментами, как число молекул, прореагировавших на активном центре в единицу времени, может быть полезным понятием, если использовать его с осторожностью. С точки зрения проблемы измерения числа активных центров, рассмотренной в §1.2.4, важно точно определить, как именно выражается Q через активные центры. Реальной мерой таких центров может быть число поверхностных атомов металла в нанесенном катализаторе, но в других случаях единственно возможной может быть оценка, базирующаяся на удельной величине поверхности по БЭТ методу. Конечно, числа оборотов (подобно скоростям) должны быть отнесены к конкретным условиям, таким как температура, начальная концентрация или начальное парциальное давление, или степень реакции.
При сравнении различных катализаторов для данной реакции или различных реакций на данном катализаторе может оказаться неудобно или невозможно сравнивать скорости при конкретной температуре, так как скорости должны измеряться при температурах, при которых они имеют обычные величины. Поэтому может оказаться целесообразным сравнивать температуры, при которых скорости имеют конкретные значения.
В реакторах с пространственно однородными концентрациями реагентов и продуктов скорости одинаковы по всей поверхности катализатора в любой момент времени. Однако в проточных интегральных реакторах скорость на каждом участке каталитического слоя меняется по длине.
Селективность
Термин "селективность", S используется для описания относительных скоростей двух или более конкурирующих реакций на одном катализаторе. Такая конкуренция подразумевает как разные реагенты, подвергающиеся одновременным превращениям, так и один реагент, участвующий в двух или более реакциях. В последнем случае S может быть определена двумя способами. Первый из них определяет избирательную селективность SF по каждому продукту по уравнению
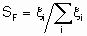
а второй определяет относительную селективность SR по каждой паре продуктов:
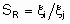
При селективности по форме, которая может наблюдаться в катализаторах с очень маленькими порами, селективность в большой степени определяется объемом или размером одного или более реагентов. На цеолитах, например, реакция алканов с линейными углеродными цепочками может идти с намного большей скоростью, чем реакция с разветвленными цепочками.
1.5.3 Уравнения скорости
Сначала рассмотрим газообразные системы, в которых все концентрации пространственно однородны и в которых реакция необратима.
Скорость x , кроме того, что она пропорциональна количеству катализатора Q, также является в общем случае функцией температуры T и концентраций ci или парциальных давлений pi реагентов, продуктов или других веществ, если они присутствуют:
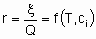
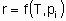
Такая форма уравнения обычно называется уравнением скорости или законом скорости. В гетерогенном катализе функция f часто записывается в форме
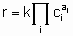
где k - константа скорости, являющаяся функцией температуры, но не концентрации, а ai (целое или дробное; положительное, отрицательное число или ноль) - это порядок реакции по компоненту i. Такая форма закона скорости называется степенным законом скорости. Однако, часто для скорости используется дифференциальная форма записи. Например, для реакции А + В → продукты уравнением скорости может быть
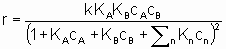
Интерпретация этого уравнения возможна с точки зрения изотерм адсорбции Лэнгмюра. Предполагается (см. §1.5.4), что оба реагента должны адсорбироваться, чтобы вступить в реакцию, и что KA и KB - это соответствующие константы равновесия адсорбции Лэнгмюра. Знаменатель учитывает конкуренцию за места между реагентами и другими веществами (растворители, яды и продукты), присутствующими в системе в концентрациях cn, с соответствующими константами равновесия адсорбции Kn. Закон скорости в таком виде можно назвать законом скорости Лэнгмюра, хотя он был введен в использование Хиншельвудом, Швабом, Хоугеном, Ватсоном и др. Такие законы скорости часто используются в применении к системам, в которых адсорбция не подчиняется изотерме адсорбции Лэнгмюра. В таких условиях законы скорости все же могут дать удобное средство сопоставления экспериментальных результатов, величины выведенных констант следует использовать с осторожностью.
Для отдельного элементарного процесса
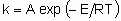
где А - это частотный фактор, а Е - энергия активации. Несмотря на то, что гетерогенные каталитические реакции редко (или даже никогда) состоят из одного элементарного процесса, такое же соотношение часто применяется для определения общей константы скорости. В таком случае, однако, А - это не частотный фактор, его следует называть предэкспоненциальным множителем, а Е - кажущейся энергией активации.
Иногда наблюдается компенсация А и Е, т.е. они изменяются в одном направлении при изменении катализатора в данной реакции или при изменении реакции на данном катализаторе. Особый случай компенсации, называемый q -правилом, имеет место, когда, по крайней мере приблизительно,

где Tq - изокинетическая температура, т.е. температура, при которой все k будут одинаковыми.
Подобные рассуждения могут быть распространены на обратимые процессы. Они также применимы и к однофазным, жидким системам. В достаточно распространенном для гетерогенных катализаторов случае, когда один реагент находится в газовой фазе, а другие реагенты и продукты - в жидкой фазе, вышеупомянутые принципы применяются непосредственно при условии, что существует равновесие массопереноса между газовой и жидкой фазами, т.е. фугитивность реагентов в газовой фазе равна их фугитивности в жидкой фазе. В таком случае степенной закон скорости для необратимой реакции в форме
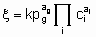
может применяться там, где величины имеют те же значения, что и раньше, за исключением того, что газообразный реагент g вводится не как ci, а как член давления с порядком ag.
Определение скорости реакции в проточной системе требует знания как скорости подачи, v, данного реагента, так и степени конверсии, x. Определение скорости подачи как количества реагента, подаваемого в единицу времени на вход в реактор, согласуется с §1.5.1. Тогда скорость реакции будет
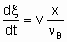
где n B - стехиометрический коэффициент реагента со степенью конверсии х. С другой стороны, можно исходить из rm, ri, и ra, а не dx /dt, определяя объемные скорости, vm, vv и va, где величины vi представляют собой скорость подачи данного реагента на единицу массы, объема или поверхности катализатора. Соотношение
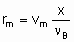
дает удельную скорость реакции или, при определенных условиях, удельную активность катализатора. Подстановка va или vv дает поверхностную скорость реакции или скорость, деленную на объем катализатора, соответственно. С другой стороны, можно использовать величины объемного времени t m, t a и t v, обратные объемным скоростям. "Время контакта" и "время пребывания" - термины, которые могут вводит в заблуждение при рассмотрении проточных систем в гетерогенном катализе, их лучше избегать.
1.5.4 Кинетические аспекты механизма
При рассмотрении механизмов удобно использовать такие понятия, как лимитирующий скорость процесс или стадия и преобладающий поверхностный интермедиат. Скорость-лимитирующий процесс определяется, как обычно в общем случае в кинетике, как такой элементарный процесс в каталитической последовательности, который не находится в равновесии, когда суммарная реакция удаляется от равновесия. Если поверхность катализатора содержит каталитические центры одного типа, данный интермедиат называют преобладающим поверхностным интермедиатом, если он покрывает намного большую часть поверхности, чем каждый из остальных интермедиатов. Конечно, нет гарантии, что скорость-лимитирующий процесс или преобладающий поверхностный интермедиат будут существовать для каждой отдельной реакции при определенном наборе условий.
Термин реакционный центр (reaction center) может относиться как к вакантным, так и к занятым каталитическим центрам (catalytic sites). Сумма поверхностных концентраций реакционных центров есть величина постоянная L. Таким образом, если группы m с поверхностной концентрацией Lm - преобладающий поверхностный интермедиат, Lm + Lv » L, где Lv - поверхностная концентрация вакантных реакционных центров.
Механизм Лэнгмюра-Хиншельвуда. Это несколько необычное применение термина "механизм" для определения относительных величин констант скоростей. В механизме Лэнгмюра-Хиншельвуда все стадии адсорбции-десорбции находятся, в основном, в равновесии, а скорость определяющей является поверхностная стадия. Такая поверхностная стадия может включать мономолекулярную реакцию одной молекулы адсорбата или реакцию двух или более молекул друг с другом на соседних местах. Там, где адсорбционные процессы идут по изотермам адсорбции Лэнгмюра, суммарная реакция будет подчиняться некоторой разновидности закона скорости Лэнгмюра (§1.5.3). Однако термин "механизм Лэнгмюра-Хиншельвуда" может относиться к ситуациям, в которых не применяются изотермы адсорбции Лэнгмюра.
1.5.5 Неоднородность каталитических центров
Для каталитической поверхности характерно то, что ее центры могут различаться по термодинамическим и кинетическим свойствам. В кинетическом описании каталитических реакций на неоднородных центрах параметр a часто используется, чтобы связать изменения энергии активации активированной адсорбции с энтальпией адсорбции
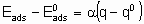
где 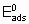 - энергия активации, а
- энергия активации, а
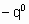 - энтальпия адсорбции на непокрытой поверхности. Eads и q относятся к поверхности с одинаковой величиной q . На практике это уравнение может применяться только в ограниченном диапазоне q . Иногда a
определяется так же, как в вышеупомянутом уравнении, но в терминах энергии активации Гиббса и адсорбции, соответственно. Термин коэффициент переноса использовался электрохимиками для выражения a в других условиях.
- энтальпия адсорбции на непокрытой поверхности. Eads и q относятся к поверхности с одинаковой величиной q . На практике это уравнение может применяться только в ограниченном диапазоне q . Иногда a
определяется так же, как в вышеупомянутом уравнении, но в терминах энергии активации Гиббса и адсорбции, соответственно. Термин коэффициент переноса использовался электрохимиками для выражения a в других условиях.
Перевод Е.Б. Никифоровой
1.3 Composition, structure and texture of catalysts
1.3.1 General terms
Catalysts may be one-phase or multiphase. In the first case, they may be composed of one substance (for example, alumina or platinum black) or they may be а one-phase solution of two or more substances. In this case, the components of the solution should be given and joined by а hyphen (for example, silica-alumina).
Support. In multiphase catalysts, the active catalytic material is often present as the minor component dispersed upon а support sometimes called а carrier. The support may be catalytically inert but it may contribute to the overall catalytic activity. Certain bifunctional catalysts (§1.2.8) constitute an extreme example of this. In naming such а catalyst, the active component should be listed first, the support second and the two words or phrases should be separated by а solidus, for example, platinum/silica or platinum/silica-alumina. The solidus is sometimes replaced by the word "on," for example, platinum on alumina.
Promoter. In some cases, а relatively small quantity of one or more substances, the promoter or promoters, when added to а catalyst improves the activity, the selectivity, or the useful lifetime of the catalyst. In general, а promoter may either augment а desired reaction or suppress an undesired one. There is no formal system of nomenclature for designating promoted catalysts. One may, however, for example, employ the phase "iron promoted with alumina and potassium oxide."
А promoter which works by reducing the tendency for sintering and loss of area may be called а textural promoter (see §1.7.3).
Doping. In the case of semiconducting catalysts, а small amount foreign material dissolved in the original catalyst may modify the rate of а particular reaction. This phenomenon is sometimes called doping by analogy with the effect of similar materials upon semiconductivity.
1.3.2 Porosity and texture
Many but not all catalysts are porous materials in which most of the surface area is internal. It is sometimes convenient to speak of the structure and texture of such materials. The structure is defined by the distribution in space of the atoms or ions in the material part of the catalyst and, in particular, by the distribution at the surface. The texture is defined by the detailed geometry of the void space in the particles of catalyst. Porosity is а concept related to texture and refers to the pore space in а material. With zeolites, however, much of the porosity is determined by the crystal structure.
An exact description of the texture of а porous catalyst would require the specification of а very large number of parameters. The following averaged properties are often used.
With respect to porous solids, the surface associated with pores may be called the internal surface. Because the accessibility of pores may depend on the size of the fluid molecules, the extent of the accessible internal surface may depend on the size of the molecules comprising the fluid, and may be different for the various components of а fluid mixture (molecular sieve effect).
When а porous solid consists of discrete particles, it is convenient to describe the outer boundary of the particles as external surface.
It is expedient to classify pries according to their sizes
(iii) pores of intermediate size are called mesopores.
The terms intermediate or transitional pores, which have been used in the past, are not recommended.
In the case of micropores, the whole of their accessible volume may be regarded as adsorption space.
The above limits are to some extent arbitrary. In some circumstances it may prove convenient to choose somewhat different values.
Pore-size distribution is the distribution of pore volume with respect to pore size; alternatively, it may be defined by the related distribution of pore area with respect to pore size. It is an important factor for the kinetic behaviour of а porous catalyst and thus an essential property for its characterisation (see §1.6).
The computation of such а distribution involves arbitrary assumptions and а pore-size distribution should always be accompanied by an indication as to the method used in its determination. The methods usually involve either or both of the following: (i) adsorption - desorption isotherms of nitrogen or other adsorptives in conjunction with а particular model for conversion of the isotherm into а pore-size distribution, (ii) data obtained by the mercury porosimeter. The isotherm gives а pore-size distribution for mesopores. The mercury porosimeter gives а distribution covering macro- pores and larger mesopores. In both cases chat is measured is, strictly speaking, not the exact volume of pores having а given pore size, but the volume of pores accessible through pores of а given size. The relationship between these two functions depends on the geometrical nature of the pore system.
The specific pore volume is the total internal void volume per unit mass of adsorbent. Some of the pore volume may be completely enclosed, and thus inaccessible to molecules participating in а catalytic reaction.
The total accessible pore volume may be measured by the amount of adsorbate at the saturation pressure of the adsorptive, calculated as liquid volume, provided the adsorption on the external surface can be neglected or can be evaluated. The accessible pore volume may be different for molecules of different sizes. А method which is not subject to the effect of the external surface is the determination of the dead space by means of а non-sorbable gas (normally helium) in conjunction with the determination of the bulk volume of the adsorbent by means of а non-wetting liquid or by geometrical measurements.
Primary particles. Certain materials widely used as catalysts or supports consist of spheroids of about 10 nm (100 Å) in diameter loosely cemented into granules or pellets. The texture of these resembles that of а cemented, loose gravel bed. The 10 nm (100 Å) particles may be called primary particles.
Percentage exposed in metallic catalysts. The accessibility of the atoms of metal in metallic catalysts, supported or unsupported, depends upon the percentage of the total atoms of metal which are surface atoms. It is recommended that the term percentage exposed be employed for this quantity rather than the term dispersion which has been frequently employed.
Pretreatment and activation. Following the preparation of а catalyst or following its insertion into а catalytic reactor, а catalyst is often subjected to various treatments before the start of а catalytic run. The term pretreatment may, in general, be applied to this set of treatments. In some cases the word activation is used. It implies that the material is converted into а catalyst or into а very much more effective one by the pretreatment. Outgassing is а form of pretreatment in which а catalyst is heated in vacuo to remove adsorbed or dissolved gas. Calcination is а term which means heating in air or oxygen and is most likely to be applied to а step in the preparation of а catalyst.
1.4 Catalytic reactors
The vessel in which а catalytic reaction is carried out is called а reactor. Many different arrangements can be adopted for introducing the reactants and removing the products.
In а batch reactor the reactants and the catalyst are placed in the reactor which is then closed to transport of matter and the reaction is allowed to proceed for а given time whereupon the mixture of unreacted material together with the products is withdrawn. Provision for mixing may be required.
In a flow reactor, the reactants pass through the reactor while the catalysis is in progress. Many variations are possible.
The catalyst may be held in а packed bed and the reactants passed over the catalyst. А packed bed flow reactor is commonly called а fixed bed reactor and the term plug-flow is also used to indicate that no attempt is made to back-mix the reaction mixture as it passes through the catalyst bed. The main modes of operation of a flow reactor are differential involving а small amount of reaction so that the composition of the mixture is approximately constant throughout the catalyst bed, or integral involving а more substantial amount of reaction such that the composition of material in contact with the final section of the catalyst bed is different from that entering the bed.
In а pulse reactor, а carrier gas, which may be inert or possibly one of the reactants, flows over the catalyst and small amounts of the other reactant or reactants are injected into the carrier gas at intervals. А pulse reactor is useful for exploratory work but kinetic results apply to а transient rather than to the steady state conditions of the catalyst.
Several alternative modes of operation may be used to avoid the complications of the changing concentrations along the catalyst bed associated with integral flow reactors and each of these has а special name. In а stirred flow reactor, effective mixing is achieved within the reactor often by placing the catalyst in а rapidly-rotating basket. If the mixing achieved in this way is efficient, the composition of the mixture in the reactor will be close to that of the exit gases.
The same result can be reached by recirculation of the gas around а loop containing а fixed bed of catalyst, provided that the rate of recirculation is considerably larger then the rate of flow in and out of the loop. Under these circumstances, а substantial conversion to products can be obtained even though conditions in the bed correspond more closely to those associated with а differential rather than with an integral reactor. Another mode of operation involves a fluidised bed in which the flow of gases is sufficient to cause the bed of finely divided particles of catalyst to behave like а fluid. In а fluidised bed, the temperature is uniform throughout, although mixing of gas and solid is usually incomplete. It has special applications in cases where the catalyst has to be regenerated, е.g., by oxidation, after а short period of use. Continuous transfer of catalyst between two vessels (one used as reactor and the other for catalyst regeneration) is possible with а fluidised system. The stirred flow and the recirculation reactors are characterised ideally by very small concentration and temperature gradients within the catalyst region. The term, gradientless reactor, may be used to include both types.
All reactors, batch or flow, may be operated in three main ways in regard to temperature. These are isothermal, adiabatic and temperature-programmed. For the last, in а batch reactor the variation of temperature with time may be programmed, or in а fixed bed reactor the variation of temperature along the length of the bed may be controlled.
When reactors are operated isothermally the batch reactor is characterised by adsorbate concentrations and other aspects of the state of the surface which are constant in space (i.е., uniform within the catalyst mass) but which change with time. In the integral flow reactor with the catalyst at steady state activity, the surface conditions are constant with time but change along the bed. In the gradientless reactor at steady state, the surface conditions are constant in space and, if the catalyst is at а steady state, with time. In the pulse reactor, the catalyst is often not in а condition of steady state, concentrations change as the pulse moves through the bed, and there may be chromatographic separation of reactants and products.
In general, if heterogeneous catalytic reactions are to be conducted isothermally, the reactor design must provide for heat flow to or from the particles of catalyst so as to keep the thermal gradients small. Otherwise, temperatures within the catalyst bed will be non-uniform. The differential reactor and the various forms of the gradientless reactors are advantageous in this regard.
The types of reactors described above can, in principle, be extended to reactions in the liquid phase although the pulse reactor has been little used in such cases.
Reactions in which one reactant is gaseous, the other is in а liquid phase, and the catalyst is dispersed in the liquid phase, constitute а special but not unusual case, for example, the hydrogenation of а liquid alkene catalysed by platinum. А batch reactor is most commonly employed for laboratory scale studies of such reactions. Mass transport from the gaseous to the liquid phase may reduce the rate of such а catalytic reaction unless the contact between the gas and the liquid is excellent (see §1.6).
1.5 Kinetics of heterogeneous catalytic reactions
1.5.1 General terms
Consider а chemical reaction
,where n B is the stoichiometric coefficient (plus for products, minus for reactants) of any product or reactant В. The extent of reaction x is defined (see §11.1 of the Manual)
,where nB is the amount of the substance В.
If rate of reaction is to have an unambiguous meaning, it should be defined as the rate of increase of the extent of reaction
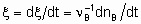
whereas the quantity dnB/dt may be called the rate of formation (or consumption) of B
То facilitate the comparison of the results of different investigators, the rates of heterogeneous catalytic reactions should be suitably expressed and the conditions under which they have been measured should be specified in sufficient detail. If the rate of the uncatalysed reaction is negligible, the rate of the catalysed reaction may be given as
.If Q, the quantity of catalyst, is in mass,
and rm is the specific rate of reaction which may be called the specific activity of the catalyst under the specified conditions. If Q is in volume,
.The volume should be that of the catalyst granules excluding the intergranular space. If Q is in area,
.where ra is the areal rate of reaction. If the total surface area of the catalyst is used, it should be preferably а ВЕТ nitrogen area. However, other types of specified areas may be employed, for ех. spic, the exposed metal area of а supported metallic catalyst. The exposed metal area is often estimated by selective chemisorption of а suitable sorptive, е.g., hydrogen or carbon monoxide.
The turnover frequency, N, (commonly called the turnover number) defined, as in enzyme catalysis, as molecules reacting per active site in unit time, can be а useful concept if employed with care. In view of the problems in measuring the number of active sites discussed in §1.2.4, it is important to specify exactly the means used to express Q in terms of active sites. А realistic measure of such sites may be the number of surface metal atoms on а supported catalyst but in other cases estimation on the basis of а ВЕТ surface area may be the only readily available method. Of course, turnover numbers (like rates) must be reported at specified conditions of temperature, initial concentration or initial partial pressures, and extent of reaction.
In comparing various catalysts for а given reaction or in comparing various reactions on а given catalyst, it may be inconvenient or impracticable to compare rates at а specified temperature since rates must be measured at temperatures at which they have convenient values. Therefore, it may be expedient to compare the temperatures at which the rates have а specified value.
In reactors in which the concentrations of reactants and products are uniform in space, the rate is the same on all parts of the catalyst surface at any specified time. In integral flow reactors, however, the rate on each element of the catalyst bed varies along the bed.
1.5.2 Selectivity
The term selectivity S is used to describe the relative rates of two or more competing reactions on а catalyst. Such competition includes cases of different reactants undergoing simultaneous reactions or of а single reactant taking part in two or more reactions. For the latter case, S may be defined in two ways. The first of these defines а fractional selectivity SF for each product by the equation
.The second defines relative selectivities, SR, for each pair of products by:
.In shape selectivity, which may be observed in catalysts with very small pores, the selectivity is largely determined by the bulk or size of one or more reactants. On zeolites, for example, the rate of reaction of alkanes with linear carbon chains may be much greater than that of those with branched chains.
1.5.3 Rate equations
Gaseous systems in which all concentrations are uniform in space and in which the reaction is irreversible will be considered first.
The rate x, besides being proportional to the quantity of catalyst, Q, is also in general а function of temperature Т and the concentrations сi or partial pressures рi of reactants, products and other substances if present:
or
The statement of this equation is commonly called the rate equation or the rate law. Frequently, in heterogeneous catalysis, the function f is of the form
.where k is the rate constant which is а function of temperature but not of concentrations and аi (integral or fractional; positive, negative or zero) is the order of the reaction with respect to component i. This form of the rate law is called а power rate law. Often, however, а rate expression of different form is used. For example, or а reaction A + B ® products, the rate equation might be
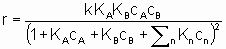 .
.This equation can be interpreted in terms Langmuir adsorption isotherms. It is assumed (see §1.5.4) that both reactants must be adsorbed in order to react and that KA and KB are the respective Langmuir adsorption equilibrium constants. The denominator allows for competition for sites between reactants and other substances (diluents, poisons and products) present in the system at concentrations cn with related adsorption equilibrium constants Кn. А rate law of this type is appropriately called a Langmuir rate law although it was made popular by Hinshelwood, Schwab, Hougen, Watson and others. Such rate laws are frequently used for systems in which the adsorptions may not obey the Langmuir adsorption isotherm. Under these circumstances, the rate laws can still provide а useful means of correlating experimental results but the values of the derived constants must be interpreted with caution.
For а single elementary process,
.where А is the frequency factor and Е the activation energy. Even though heterogeneous catalytic reactions rarely if ever proceed by а single elementary process, the same relation often applies to the overall rate constant. In such a case, however, А is not а frequency factor but should be called the preexponential factor and Е should be called the apparent activation energy.
Sometimes А and Е exhibit compensation, i.е., they change in the same direction with change in catalyst for а given reaction or with change in reaction for а given catalyst. А special case of compensation called the -rule occurs when, at least approximately,
..This equation can be interpreted in terms Langmuir adsorption isotherms. It is assumed (see §1.5.4) that both reactants must be adsorbed in order to react and that KA and KB are the respective Langmuir adsorption equilibrium constants. The denominator allows for competition for sites between reactants and other substances (diluents, poisons and products) present in the system at concentrations cn with related adsorption equilibrium constants Кn. А rate law of this type is appropriately called a Langmuir rate law although it was made popular by Hinshelwood, Schwab, Hougen, Watson and others. Such rate laws are frequently used for systems in which the adsorptions may not obey the Langmuir adsorption isotherm. Under these circumstances, the rate laws can still provide а useful means of correlating experimental results but the values of the derived constants must be interpreted with caution.
For а single elementary process,
.where А is the frequency factor and Е the activation energy. Even though heterogeneous catalytic reactions rarely if ever proceed by а single elementary process, the same relation often applies to the overall rate constant. In such a case, however, А is not а frequency factor but should be called the preexponential factor and Е should be called the apparent activation energy. .
Sometimes А and Е exhibit compensation, i.е., they change in the same direction with change in catalyst for а given reaction or with change in reaction for а given catalyst. А special case of compensation called the q-rule occurs when, at least approximately,
.where Т is the isokinetic temperature, the temperature at which all k's would be identical.
These considerations can be extended to reversible processes. They also apply to single phase, liquid systems, For the case, rather common in heterogeneous catalysts, in which one reactant is in а gas phase and the others and the products are in а liquid phase, application of the principles given above is straightforward provided that there is mass transfer equilibrium between gas phase and liquid phase, i.e., the fugacity of the reactant in the gas phase is identical with its fugacity in the liquid phase. In such case, а power rate law for an irreversible reaction of the form
.may apply where the quantities have the same significance as before except that the gaseous reactant g is omitted from the ci's and entered as а pressure term with order аg.
The determination of rate of reaction in а flow system requires knowledge both of the feed rate, v, of а given reactant and of the fraction converted, x. The definition of feed rate as the amount of reactant fed per unit time to the inlet of the reactor is consistent with §;1.5.1. The rate of reaction is then given by
.where n Bis the stoichiometric coefficient of the reactant of which the fraction x is converted. Alternatively, one may proceed from rm, ri, and ra rather than dx /dt by defining the space velocities, vm, vv, and va where the vi's represent the rate of feed of the given reactant fed per unit mass, volume or surface area of the catalyst. The relation,
.gives the specific rate of reaction or, under specified conditions, the specific activity of the catalyst. Substitution of va or vv gives the areal rate of reaction or the rate divided by volume of the catalyst, respectively. Alternatively, space times, t m, t a, and t v, the reciprocals of the space velocities, may be used. "Contact time" and "residence time" are terms which may be misleading for flow systems in heterogeneous catalysis and should be avoided.
1.5.4 Kinetic aspects of mechanism
Of general convenience in the treatment of mechanisms are the notions of rate determining process or step and most abundant surface intermediate. The rate determining process is defined, as is usual in kinetics in general, as that single elementary process in the catalytic sequence which is not in equilibrium when the overall reaction is significantly displaced from equilibrium. If the surface of а catalyst has one set of catalytic sites, а particular intermediate is said to be the most abundant surface intermediate if the fractional coverage by that intermediate is much larger than coverages by the other intermediates. Of course, there is no guarantee that either а rate determining process or а most abundant surface intermediate will exist for any particular reaction under а particular set of conditions.
The term reaction centre may be used to include both vacant and occupied catalytic sites. The sum of the surface concentrations of reaction centres on the surface of а catalyst is а constant L. Thus, if species m at а surface concentration Lm is the most abundant surface intermediate,
Lm + Lv »
L, where Lv is the surface concentration of vacant reaction centres.
Langmuir-Hinshelwood mechanism. This represents a somewhat anomalous use of the term mechanism to specify relative magnitudes of rate constants. In a Langmuir-Hinshelwood mechanism, all adsorption-desorption steps are essentially at equilibrium and a surface step is rate determining. Such a surface step may involve the unimolecular reaction of a single adsorbate molecule or the reaction of two or more molecules on adjacent sites with each other. Where the adsorption processes follow Langmuir adsorption iso therms, the overall reaction will follow some kind of a Langmuir rate law (§1.5.3). However, the term Langmuir-Hinshelwood mechanism may cover situations in which Langmuir adsorption isotherms do not apply.
1.5.5 Non-uniformity of catalytic sites
A characteristic of a catalytic surface is that its sites may differ in their thermodynamic and kinetic properties. In the kinetic description of catalytic reactions on non-uniform surfaces, a parameter a is frequently used to connect changes in the activation energy of activated adsorption with the enthalpy of the adsorption
.where 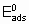 is the energy of activation and -q0 is the enthalpy of adsorption on the uncovered surface. Eads and q apply to the surface with the same value of a
. In practice the equation may apply only over a restricted range of q
. Sometimes a
is defined as in the equation above but in terms of Gibbs energies of activation and adsorption, respectively. The name transfer coefficient has been used by electrochemists to represent a
in another related situation.
is the energy of activation and -q0 is the enthalpy of adsorption on the uncovered surface. Eads and q apply to the surface with the same value of a
. In practice the equation may apply only over a restricted range of q
. Sometimes a
is defined as in the equation above but in terms of Gibbs energies of activation and adsorption, respectively. The name transfer coefficient has been used by electrochemists to represent a
in another related situation.
Уважаемые коллеги!
Представляем Вашему вниманию информацию по различным вопросам, связанным c третьим тематическим направлением Шестой рамочной программы FP6-NMP:
- 1 апреля 2005 г. в Институте кристаллографии РАН состоялся информационный день "Try to give more information to encourage the participation and to improve the cooperation", ставший первой презентацией в России 3-го тематического приоритета Шестой рамочной программы Евросоюза "Нанотехнологии и нанонауки, интеллектуальные многофункциональные материалы, новые устройства и производственные процессы" (FP6-NMP).
Некоторые выступления представлены на сайте
Национального контактного центра России:
http://www.fp6-nano.com/infoday/infoday.html
В них содержится полезная информация о правилах участия российских организаций в Шестой рамочной программе, об используемой Еврокомиссией системе оценки представленных на конкурсы проектов, о мобильности ученых и т.д.
- В рамках Интернет-портала "Наука и технологии в России и других странах СНГ" создана новая база данных по проектам иностранных партнёров Международного научно-технического центра (МНТЦ) для налаживания сотрудничества с учёными СНГ в высокотехнологичных областях.
Подробная информация на сайте: http://tech-db.istc.ru/ISTC/sc.nsf/html/stn-database.htm
- 14-15 апреля в Брюсселе проходил семинар "Research - Training in Nanosciences and Nanotechnologies: Current Status and Future Needs", организованный Директоратом по научным исследованиям Еврокомиссии. В семинаре участвовали представители академической науки и промышленности из 27 европейских стран, США и Азии. Целью семинара было обсуждение различных аспектов образования и научных исследований для об еспечения прогресса в новых технологиях в мире и, в частности, в Европе. Подробная информация о семинаре и все презентации представлены на сайте: http://www.cordis.lu/nanotechnology/src/educationworkshop.htm
- Европейская Комиссия представила широкой общественности планы, связанные с 7-й Рамочной программой (FP7), рассчитанной на семь лет (2007-2013 гг.), бюджет которой составит 73 млрд. e. С инициативами Еврокомиссии по FP7 можно познакомиться на сайте www.cordis.lu/fp7/
- 7 июня 2005 г. Еврокомиссией принят план дальнейших действий в области нанотехнологий и нанонауки ("Nanosciences and Nanotechnologies", N&N)на 2005-2009 гг. Документ отражает совокупность мер, обеспечивающих сохранение Европой лидирующих позиций в области нанотехнологий. С документом можно ознакомиться детально на сайте: http://www.cordis.lu/nanotechnology/src/communication.htm
- Создан сайт успешных проектов Шестой рамочной программы с российскими участниками. Адрес сайта: http://www.singleimage.co.uk/russia-in-ist/site.php?PHPSESSID=422faf45990442171ae7cacd225b4297
- В 2005 г. Физико-технический институт им. Иоффе организует ряд международных конференций по различным узкотематическим направлениям в области нанотехнологий. Подробная информация может быть получена на страничке сайта института (Events 2005): http://www.ioffe.ru/evpti_05.html
- Новые предложения относительно поиска партнеров для участия в проектах FP6-NMP Вы найдете в разделе "Поиск партнёров" сайта:
http://www.fp6-nano.com/partners.php
- ЕвроНаноФорум-2005 "Нанотехнологии и здравохранение для граждан Европейского Союза в 2020 г." пройдёт 6-9 сентября 2005 г. в Эдинбурге (Шотландия). Подробная информация на сайте www.euronanoforum2005.org.
- Вторая Европейская всеобщая научная конференция состоится 14-15 ноября 2005 г. в Брюсселе. Организатор конференции - Директорат по науке Европейской Комиссии.
Смотрите сайт: http://europa.eu.int/comm/research/conferences/2005/cer2005/pdf/cer_announcement_en.pdf
- Европейская Комиссия начинает исследование эффективности научно-технологических разработок в секторе микро/наноэлектро-ники с целью повышения конкурентоспособности Европейской промышленности.
С целью привлечения к исследованиям самого широкого спектра организаций разослана анкета компаниям, научным лабораториям, менеджерам, экспертам, политикам и т.д.
На вопросы предлагаемой анкеты могут ответить все желающие. Ответы носят конфиденциальный характер и не будут использованы вне рамок данного исследования.
Любые вопросы относительно этого исследования можно задавать по адресу: Team@NanoElectronics4Europe.org.
Анкету можно найти на сайте:
http://www.nanoelectronics4europe.org/survey/index.php?sid=2&token=0921705331
- Открыта программа грантов для молодых ученых, участвующих в летних школах ИНТАС (сайт: <http://www.intas.be/index.asp?s=2_5&uid>)
Ее цель - стимулировать участие молодых ученых из России и стран СНГ в летних школах ИНТАС для налаживания контактов с учеными из стран Евросоюза и более активного вовлечения в международное научное сообщество.
Более подробная информация на сайте:
<http://www.intas.be/> в "Funding Opportunities"
- Международный нанотехнологический конгресс пройдет 1-3 ноября 2005 г. в Сан-Франциско. Подробная информация на сайте: nanotechcongress.com.
- страничка на сайте CORDIS http://www.cordis.lu/nanotechnology/src/pressroom_projects.htm
представляет список проектов FP6-NMP в области нанотехнологий со ссылками на новые статьи, успешные истории и описания проектов.
Более общую информацию о проектах можно найти на сайте:
FP6 CORDIS projects database.
- основная библиотека проектов Шестой рамочной программы в области нанотехнологий представлена на странице : http://europa.eu.int/comm/research/fp6/projects.cfm?p=3&pmenu=of
- Вниманию российских организаций, заинтересованных в сотрудничестве с европейскими партнерами предлагается анкета по поиску партнеров "PARTNER SEARCH FORM FOR EUROPEAN RTD PROJECTS", разработанная австрийскими коллегами из FFG
(FFG - Austrian Research Promotion Agency, Division for European and International Programmes (EIP)).
Контактная информация:
FFG, Donau-City-Str. 1, A-1220 Vienna, Austria
Robert Worel,
Phone: +43 (0)5 7755 4611,
Fax: +43 (0)5 7755 94011,
E-mail: robert.worel@ffg.at
Анкету Вы можете найти на сайте:
http://fp6-nano.com/contacts.php
- Не закрыт конкурс по традиционным инструментам STREP и SSA:
FP6-2004-NMP-TI-4
http://fp6.cordis.lu/nmp/call_details.cfm?CALL_ID=182
Дата закрытия этого конкурса 15 сентября 2005 г., 17:00 (время брюссельское)
Имея как минимум 3 партнеров на Западе, можно даже рискнуть подготовить конкурентоспособный проект для конкурса.
ACS Award in Industrial Chemistry
ponsored by the ACS Division of Business Development & Management
Edwin A. Chandross is "one of the world's premier industrial chemists," according to former colleague Valerie J. Kuck. During his tenure at Bell Laboratories - formerly part of AT&T and nowpart of its successor, Lucent Technologies - "Ed carried out an exceptional and diverse research and development program aimed at the design and implementation of organic materials and chemical structures for advanced telecommunication technologies," Kuck adds. Before she retired from Lucent in 2001, she was a member of technical staff.
Chandross, 70, earned a B.S. in chemistry in 1955 at Massachusetts Institute of Technology, and an M. A. in 1957 and a Ph.D. in I960, both in chemistry, at Harvard University.
Chandross says he joined Bell Labs in 1959 as a member of technical staff after the company "made me an offer I couldn't refuse." The lab, which was doing little fundamental chemical research at the time, invited him to come and set up his own research program. "Jobs like that hardly existed then, and they certainly don't exist today" Chandross says. He and five other newly minted organic chemistry Ph.D.s joined Ed Wasserman, who was already on staff (and who was to become ACS president in 1999). The group Сwas one of several that put Bell Labs on the map as a real chemistry powerhouse."
"It was a very collegial group," Chandross recalls. "We would comment on and review each others' work in a very friendly style." The collaborative style extended well beyond the circle of chemists. "Multidiscipli-nary work was a hallmark of Bell Labs, and I enjoyed collaborating with top-notch scientists in many fields," Chandross says.
Chandross flourished, conducting both fundamental and applied research and often bringing his own experience to bear on problems that stumped people in other departments. "It was fun to take something as prosaic as how to remove polymers thoroughly from glassware" - a technique he picked up as a graduate student - "and turn it into an industrial process used in splicing optical fibers," he says.
Holder of approximately 60 patents, Chandross has had a hand in the development of many products and technologies, including some familiar to the public. For example, the light stick products made by American Cyanamid were based on his discovery that hydrogen peroxide reacts with an oxalate to produce an intermediate that can cause many different compounds to fluoresce.
His other advances include the discovery of electron-transfer chemilumi-nescence; the study of excimer properties; the development of holographic storage materials; a simple photochemical technique to reduce precursor impurities, which yielded lower loss optical fibers; early development of deep ultraviolet photoresists; and new techniques for making thin-film optical waveguides, gratings, and solid-state dye lasers. After almost 42 years with Bell Labs and Lucent, Chandross retired as director of Lucent's materials chemistry department, though he still does part-time work for the firm. In 2002, he started Materials Chemistry LLC, a consulting firm in Murray Hill, N.J.
Chandross has served on advisory boards for MIT; the University of California, Los Angeles; and Northwestern University, as well as several editorial boards including Chemical Reviews and the Journal of the American Chemical Society. Chandross is a principal editor of the Journal of Materials Research and a fellow of the American Association for the Advancement of Science.
The award address will be presented before the Division of Business Development & Management.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / JENUARY 3, 2005
ACS Award in Colloid & Surface Chemistry
Sponsored by Procter & Gamble
As a 17-year veteran professor of the University of California, Berkeley, editor of Nano Letters, and the scientific founder of both NanoSys and Quantum Dot Corp., you might expect at least a little bit of hubris from 45-year-old A. Paul Alivisatos. But with his softspoken manner, Alivisatos couldn't be more easygoing about his remarkable career.
Perhaps Alivisatos' humility comes from his midwestern and Greek roots. A Chicago native, Alivisatos moved to Greece with his family when he was 10. He returned to his hometown seven years later to pursue his undergraduate degree in chemistry at the University of Chicago. Alivisatos comes from a family of doctors, and he admits that he originally intended to go into the family business until he fell under chemistry's spell.
"I liked chemistry much more than I liked medicine," he says. "I liked that level of explanation." Medicine's loss became chemistry's windfall, and Alivisatos continued his studies at UC Berkeley. There, he earned his Ph.D. in physical chemistry under the guidance of Charles Harris.
"When I finished my Ph.D., I got very interested in electronic materials, and I wanted to go to Bell Labs," Alivisatos says. There, he undertook postdoctoral research with his mentor, Louis E. Brus. This research led him to the study of nanocrystals, an area that combined his interests in chemistry and electronic solids.
In 1988, Alivisatos returned to Berkeley as an assistant professor and today is the Chancellor's Professor of Chemistry & Materials Science at the university
Interdisciplinary work in the synthesis, characterization, and application of colloidal nanocrystals has earned Alivisatos a reputation as a nanotechnology pioneer. He has devised methods to reproducibly synthesize nanocrystals with novel shapes, such as tetrapods and egg yolks. Alivisatos also demonstrated that it is possible to tune the properties of an inorganic solid in colloidal form by changing the solid's size. Several of his inventions are beginning to bear fruit in the marketplace: Paint on solar cell materials and quantum dot biological sensors born in the Alivisatos lab are currently being commercialized in the U.S. and Japan.
"Alivisatos is one of the founders of this field of chemistry," says colleague JohnT States Jr., a chemistry professor at the University of Pittsburgh. "He has played a key role in the development of the science and technology of colloidal nanocrystals, with work on every aspect of these important materials, ranging from synthesis to spectroscopy to thermodynamics to assembly and finally to application."
"Attaching colloidal semiconductors to DNA or oligonucleotides has been a major advance in the field of biosensors that would not have been possible without the work of Alivisatos," adds UC Berkeley chemistry professor Gabor A. Somorjai.
Not all of Alivisatos' contributions are confined to the laboratory He had a hand in crafting the original proposal for the National Nanotechnology Initiative, the past decade's single largest increase in government funding for the sciences outside of the health domain. And as the founding editor-in-chief of Nano Letters, he has nurtured a forum for scientific publication in the colloid chemistry community and beyond.
Naturally, Alivisatos speaks of his success with his characteristic modesty. "I'm lucky to have very good support," he says. "I have to give a lot of credit to my wife, Nicole."
The award address will be given before the Division of Colloid & Surface Chemistry.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / JENUARY 3, 2005
2005 Bower Award and Prize for Achievement in Science
Henri B. Kagan, Ph.D.
Professor Emeritus
Laboratoire de Synthese Asymetrique
Institut de Chimie Moleculaire et des Materiaux d'Orsay
Universite Paris-Sud
Orsay, France
The 2005 Bower Award and Prize for Achievement in Science has been presented to Henri B. Kagan for his seminal discovery of fundamental chemical principles that explain the impact of catalyst shape on its effectiveness in controlling chemical reactions, thus greatly simplifying the manufacture of pharmaceutically important compounds.
Henri Kagan is widely recognized as a pioneer in the field of asymmetric catalysis. His discoveries have had far-reaching impact on the pharmaceutical industry. Dr. Kagan studied at the Sorbonne in Paris before receiving his Ph.D. from the College of France in 1960. After a nearly 40-year career at the Universite Paris-Sud in Orsay, France, he now serves as an emeritus professor. His career has spanned the world, and he continues to be an active visiting lecturer, author, and enthusiastic mentor to young chemists.
Dr. Kagan has received the Silver Medal of the French National Scientific Research Center, the Prelog Medal, the August-Wihelm-von Hoffman Medal, the Nagoya Medal of Organic Chemistry, the Tetrahedron Prize for Creativity in Organic Chemistry, the Wolf Prize in Chemistry, the Grand Prix de la Fondation de la Maison de la Chimie, the Chevalier de la Legion d'Honneur, the JSPS Award for Eminent Scientists, and The Ryoji Noyori Prize. He was elected to the French Academy of Sciences and is an honorary member of, or doctor honoris causa from several institutions.
Applied Catalysis A: General
Volume 286, Issue 1 Ч 26 MAY 2005
Magnetite nanotubes
Carbon nanotubes have been the rage of materials science because of their seemingly unlimited applications. But inorganic nanotubes could have diverse applications as well, including some that take advantage of the magnetic properties of inorganic materials. Researchers are just getting up to speed on how to make magnetic nanotubes, and in one of the latest efforts, electrical engineer Chongwu Zhou of the University of Southern California and his colleagues report the first single-crystalline magnetite (Fe3O4) nanotubes [J. Am. Chem. Soc., 127, 6 (2005)]. The team used a three-step process that begins by growing magnesium oxide nanowires followed by pulsedlaser deposition of a magnetite layer to give MgO/Fe3O4 coreshell nanowires, a method Zhou's group recently reported [Nano Lett., 4, 2151 (2004)]. Finally, the inner MgO core is etched out by a solution of ammonium sulfate, leaving the 30-nm-diameter Fe3O4 nanotubes. The magnetite tubes could serve as tunable nanofluidic channels or in magnetic data storage applications, Zhou says.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / JENUARY 3, 2005
Making olefins from soybeans
Catalytic method converts soy-based biodiesel to valuable chemicals
Vegetable gardens may seem like unlikely places to look for olefins. But according to a new study the leafy patch in the backyard may be an ideal spot to collect renewable feed materials. Researchers in Minnesota have shown that olefins and olefinic esters, which are central to polymer production, can be produced from vegetable-oil-derived biodiesel using methods that are more environmentally friendly than conventional methods.
Worldwide, some 300 billion lb of olefins are produced annually - mainly from ethane and other light alkanes - through an energy-intensive process known as steam cracking. According to some estimates, nearly one-third of all pollution emitted by chemical plants is attributed to nitrogen oxides and unburned hydrocarbons released in the flames needed to drive the high-temperature process.
A less polluting and less energy-demanding method for making olefins - and one that also shifts the chemical industry's dependence on petroleum to renewable feed sources - would be an improvement over today's processes. The latest work on biodiesel suggests that such a process may be feasible.
At the University of Minnesota, Twin Cities, Lanny D. Schmidt, a professor of chemical engineering and materials science, and graduate student Ramanathan Subramanian have shown that soy-based biodiesel (a mixture of methyl esters derived from vegetable oils) can be oxidized to valuable olefins and olefinic esters efficiently and fairly selectively The reaction is conducted in an autothermal catalytic reactor, in which heat is supplied by the exothermic oxidation reactions, not by external heaters [Angew. Cbem. Int. Ed, 44, 302 (2004)].
To carry out the oxidation process, the Minnesota group uses an automotive fuel injector to spray droplets of biodiesel, which consists of methyl oleate, methyl linoleate, and related compounds, onto the walls of the reactor where the droplets vaporize. A mixture of the organic material and air is then passed over a catalyst that contains a few percent of rhodium and cerium supported on alumina.
By adjusting the ratio of biodiesel to oxygen (C/O) in the feed stream, the team is able to control the oxidation process and reactor conditions, such as catalyst temperature, and thereby tune the product distribution. For example, at a C/O ratio of roughly 1.3, the reaction yields about 25 % ethylene and smaller concentrations of propylene, 1-butene, and 1-pentene. In contrast, at a C/O ratio of 0.9, the product stream consists mainly of hydrogen and CO.
The team notes that C2 to C5 products consist almost exclusively of olefins, whereas longer chain products include olefins and olefinic esters. The researchers report that at all C/O ratios, the process yields less than 13 % CO2 (an unwanted product). They add that the catalyst remains stable and resists deactivation by carbon buildup even under extreme con-ditions.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / JENUARY 3, 2005
MS imaging methods combined
A new study combines the imaging capabilities of secondary ion mass spectrornetry (SIMS) with sample preparation techniques used in matrix-assisted laser desorption ionization (MALDI) MS. The technique, known as matrix-enhanced (ME) SIMS, provides a larger mass range than normal SIMS imaging can analyze and better spatial resolution than MALDI imaging can achieve. Sander R. Piersma and coworkers at the Institute for Atomic & Molecular Physics and Free University Amsterdam employed the standard MALDI matrix 2,5-dihydroxybenzoic acid and SIMS with an indium ion gun to obtain images of cholesterol and a neu-ropeptide called APG Warnide in tissue slices of a freshwater snail [Anal. Chem., 77, 735 2005)]. ME-SIMS increases the mass range to which SIMS can be applied, allowing the higher spatial resolution of SIMS to be applied to peptides in tissue samples.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / JENUARY 17, 2005
Greener silica
Hollow spheres are prepared with an emulsion of CO2 droplets in water
A new method for synthesizing mesoporous shells of silica combines surfactant templating and supercritical fluid processing.
"We have managed to use supercritical CO2 as a benign internal phase and swelling agent in the formation of hollow spheres of silica with large mesopore wall structures," says Robert Mokaya, a reader in materials chemistry who led the University of Nottingham, England, group that developed the technique [Chem. Common., published online Dec. 3, http://xlink.rsc.org/?doi=10.1039/b413820a].
"Hollow spheres of mesoporous silica are likely to find application in catalysis and drug delivery and as hosts for occlusion compounds in advanced composite materials," Mokaya notes. "With our technique, we are able to control both the pore size and morphology of the mesoporous silica by varying the pressure of the supercritical fluid."
The Nottingham chemists prepare the spheres, which have average pore diameters of 10 nm, by adding tetraethyl orthosilicate to an aqueous solution of a tri-block copolymer: poly(ethyleneoxide)-poly(propylene oxide)-poly(ethylene oxide). They then heat and pressurize the mixture in CO2 in an autoclave to form an emulsion of CO2 droplets in water. The copolymer acts as a surfactant at the CO2/water interface. After pressure is reduced, the product - a white powder - is separated from die mixture by filtration. Organic material is removed by heating the powder in air to 500 °C.
Other emulsion-templating methods for preparing hollow spherical materials are more complicated and less environmentally friendly, Mokaya points out. They typically employ large quantities of water-immiscible oil or an organic solvent, such as 1,3,5-trimethyl-benzene, as an internal phase.
"Our synthesis method might be extended to mesostructured hollow spheres of other (nonsilica) metal oxides or even nonoxide frameworks," Mokaya suggests. "The challenge here is to find suitable precursors from which to construct the nonsilica frame-works.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / DECEMBER 20, 2004
From benzene to picolinic acid
Japanese researchers have come up with a practical method to prepare picolinic acid-containing aromatic compounds. Such compounds are rare in nature and difficult to prepare by conventional synthesis. If readily available, they could be used as starting materials for pharmaceuticals, agro-chemicals, and other useful compounds. The team, led by Kazutoshi Shindo at Japan Women's University, Tokyo, incorporated the genes for biphenyl catabolism into Esch-erichia coli. When the reengineered bactria are grown in the presence of aromatic compounds containing two benzene rings, the bacteria transform one of the rings into picolinic acid (pyridine-2-car-boxylic acid) [J. Am. Chem. Soc., 126, 15042 (2004)]. For example, the compound shown is the product from 7-hydroxyflavanone. The researchers speculate that the expected cleavage products of the catabolic enzymes are unstable in the culture medium and are quicklyconverted to picolinic acid by incorporating ammonia and undergoing ring closure. Using II different starting materials, the team achieved bioconversion; yields ranging from 13 % for 3-phenylindanone to 84 % for 7-hydroxyflavanone.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / NOVEMBER 8, 2004
Catalysis with serial ligands
Two ligands play independent roles in bringing about different steps of a catalytic cycle, scientists have observed. The phenomenon, dubbed serial ligand catalysis, maybe occurring widely, but the work of M. Christina White and coworkers at Harvard University may be the first to explicitly demonstrate it (J. Am. Cbem. Soc., published April 21, dx.doi.org/ 10.1021/ja0500198). They obtained evidence for serial ligand catalysis from a mild and highly selective palladium-catalyzed allylic oxidation of terminal olefins. The data are consistent with a palladium-phenyl vinyl sulfoxide complex promoting the initial cleavage of the allylic carbon-hydrogen bond followed by a palladium-benzo-quinone complex promoting the functionalization of the allylic carbon. The conventional approach to homogeneous catalysis of one-metal/one-ligand combination may be inefficient when a reaction involves several product-forming steps that impose different demands on the metal, White says. "One-metal/multiple-ligand combinations may result in uniquely mild and selective solutions."
HTTP://WWW.CEN-ONLINE.ORG
С & EN / may 2, 2005
Fuel cell clean: water, makes hydrogen to boot
A prototype microbial fuel cell designed to generate a steady stream of electricity as it cleans wastewater now has been modified to produce hydrogen instead (Environ. Set. Technol., published April 22, dx.doi. org/10.1021/es050244p). Brace E. Logan and Hong Liu at Penn State University originally designed their flow-through fuel cell to use bacteria living on the carbon anode to oxidize organic matter in wastewater. Hydrogen ions and electrons generated by the oxidation combine at the cathode with O2 from air to form water and generate electricity. Bacteria have a "fermentation barrier" that limits their ability to completely degrade carbohydrates to CO2 and H2, but the Penn State researchers determined that excluding O2 from the system and applying an additional 0.25 V to the circuit could overcome the barrier. The modified fuel cell efficiently generates H2 from acetic acid in the lab, but any type of organic matter in wastewater would work. The researchers believe their fuel cell could supply enough H2 for energy production to significantly offset the cost of wastewater treatment.
HTTP://WWW.CEN-ONLINE.ORG
С & EN / may 2, 2005
Invitation
All scientists and engineers working on synthesis, characterization and applications of carbon materials in catalysis are invited to participate in the II International Symposium on Carbon for Catalysis - CarboCat-II, which will be held in St. Petersburg, Russia on July 12-14, 2006.
|
August 7-12, 2005 17th International Symposium on Plasma Chemistry Toronto, Canada |
Prof. Javad Mostaghimi |
|
August 13-21, 2005 IUPAC 43rd General Assembly Beijing, China |
IUPAC Secretariat |
|
August 14-18, 2005 11th International Symposium on Novel Aromatic Compounds (ISNA-11) St.John's, Newfoundland, Canada |
Dr.Graham Bodwell |
|
August 14-19, 2005 IUPAC 40th Congress Beijing, China |
Prof. Xibai Qiu |
|
August 21-25, 2005 International Conference on Solution Chemistry Portoroz, Slovenia |
Prof. Vojko Vlachy |
|
August 28-September 1, 2005 7th European Congress on Catalysis Sofia, Bulgaria |
Gabriele Centi |
|
August 30-September 3, 2005 European Science Education Research Association "Contributions of Research to Enhancing Students' Interest in Learning Science" Barcelona, Spain |
Dr.Roser Pinto |
|
September 3-8, 2005 16th FECHEM Conference on Organometallic Budapest, Hungary |
Ms Panna Korispataky |
|
September 4-8, 2005 European Corrosion Congress Lisbon, Portugal |
Mr.Vitor Alves |
|
September 5-9, 2005 International Symposium Recent Advances in Catalysis Rennes, France |
Dr. E. Mignard |
|
September 11-14, 2005 6th International Workshop on Catalytic Combustion Isle of Ischia, Italy |
Prof. Pio Forzatti |
|
September 13-15, 2005 Eastern Biofuels Conference & Expo Warsaw, Poland |
Web site: www.easternbiofuels.com |
|
September 15-16, 2005 International Symposium on CO2 "Reduction of emissions and geological storage of CO2: innovation and industrial stakes" Paris, France |
Patricia Fulgoni E-mail: patricia.fulgoni@ifp.fr |
|
Сентябрь 20-24, 2005 IV Всероссийская научно-практическая конференция "Проблемные вопросы утилизации смесевых твердых топлив, отходов и остатков жидких ракетных топлив в элементах ракетно-космической техники" Бийск, Россия |
Марьяш Виктор Иосифович |
|
Сентябрь, 2005 Международная конференция "Современные проблемы асимметрического синтеза" Пермь, Россия |
Институт технической химии |
|
Сентябрь, 2005 XVII Симпозиум "Современная химическая физика" Туапсе, Россия |
Институт химической физики |
|
October 4-8, 2005 1st Europe Conference on Chemistry for life Sciences Rimini, Italy |
Fax: +39 0547 382348 |
|
October 9-13, 2005 32nd International Quebec City, Canada |
FACSS National Office |
|
Октябрь 11-13, 2005 II Конференции с Уфа, Россия |
Соболева Татьяна Валериановна |
|
October 16-19, 2005 2nd International Conference on "Structured Catalysts and Reactors" Delft, Netherlands |
Prof. Jacob A. Moulijn |
|
October 17-21, 2005 3rd Asia-Pacific Symposium on Radiochemistry (APSORC'05) Beijing, China |
Prof.Z.F.Chai |
|
Октябрь 18-21, 2005 XVIII Международная научно-техническая конференция "Химические реактивы, реагенты и процессы малотоннажной химии" Минск, Беларусь |
Михайловский ёрий Кимович Минск, Беларусь |
|
October 19-21, 2005 14th Argentine Catalysis Congress Santa Fe, Argentina |
E-mail: capesteg@fiqus.unl.edu.ar |
|
Октябрь, 2005 VII Международный Фрумкинский симпозиум "Кинетика электродных процессов" Москва, Россия |
Институт электрохимии |
|
October 30-November 4, 2005 AIChE Annual Meeting |
Meetings Department |
|
November 14-17, 2005 11th Instrumental Analysis Conference and Exhibition Barcelona, Spain |
E-mail: info@jai2005.com Web site: http://www.jai2005.com |
|
November 22-25, 2005 2nd European Hydrogen Energy Conference (EHEC 2005) Zaragoza, Spain |
E-mail: info@ehec.info |
|
December 4-6, 2005 Asia Biofuels Conference Manila, Philippines |
Web site: www.asiabiofuels.com, http://www.biofuelsconferences.com/call.cfm |
|
December 14-16, 2005 International conference "Research Advances in Rational Design of Catalysts and Sorbents" IFP-Lyon, France |
Frederique Leandri |
|
December 15-20, 2005 International Symposium "Preparation of New Functional Catalysts and Photocatalysts: Theoretical Analysis, Characterizations, Reactivities and Applications" (PACIFICHEM 2005) Honolulu, Hawaii, USA |
Prof. Alex Bell |
|
2006 Euchem Conference on Organic Free Radicals Bergen, Norway |
E-mail: Lars.Engman@kemi.uu.se |
|
January 5-6, 2006 Metals and Radicals in Biology: London, United Kingdom |
Jon McMaster |
|
January 7-10, 2006 Nanoscience with Nanocrystals Grenoble, France |
Andrey Rogach |
|
January 16-18, 2006 5th Middle East Refining and Petrochemical Conference & Exhibition (PETROTECH 2006) Bahrain |
Khalida M. Al-Dalama |
|
January 22-27, 2006 9th International Conference on Solar Energy and Applied Photochemistry Cairo/Giza, Egypt |
Prof.Dr. Sabry Abdel-Mottaleb |
|
February 6-8, 2006 8th International Symposium on Advances in York, United Kingdom |
Robert Smits |
|
February 8-10, 2006 9th International York, United Kingdom |
Robert Smits |
|
March 5-10, 2006 Gordon Research Conference on New Antibacterial Discovery & Development Ventura, CA, USA |
Trevor Trust |
|
April 5-7, 2006 Materials Congress 2006 London, UK |
Lisa Bromley The Institute of |
|
April 2006 Central Biofuels Conference & Expo |
100 S Dakota Avenue |
|
April 19-21, 2006 9th Dalton Disscussion Manchester, United Kingdom |
RSC Conferences |
|
April 23-28, 2006 Marianske Lazne, |
E-mail: radchem@fjfi.cvut.cz |
|
April 24-26, 2006 133th Faraday Discussion "Chemical Evolution of the Universe" St Jacut de la Mer, Brittany , France |
RSC Conferences |
|
May 28-June 1, 2006 3rd International Tokyo, Japan |
Hiro Nishide |
|
June 4-8, 2006 International Symposium on Environmental Hamburg, Germany |
Marianne Frei |
|
June 11-14, 2006 8th Hightway and Urban Cyprus |
Alexandra Priatna |
|
June 11-14, 2006 Process Development Palm Springs, CA, USA |
Dr. Lionel O'Young |
|
June 11-15, 2006 16th International Conference on Organic Synthesis Merida, Yucatan, Mexico |
Dr. Eusebio Juaristi |
|
June 21-25, 2006 6th International |
Prof. Maciej Jarosz |
|
Summer 2006 Eastern Biofuels Conference & Expo Budapest, Hungary |
Web site: www.biofuelsconferences.com |
|
July 9-14, 2006 16th European Symposium on Computer Aided Process Engineering Garmisch-Partenkirchen, Germany |
Ms. Claudia Martz |
|
July 10-12, 2006 134th Faraday Discussion "Atomic Transport and Defect Phenomena in Guildford , United Kingdom |
RSC Conferences |
|
July 22-27, 2006 19th International Orleans, France |
Dwayne Heard |
|
July 30 - August 2, 2006 International Symposium on Zeolites and Microporous Crystals (ZMPC2006) Yonago, Japan |
Prof. Dr. Naonobu Katada |
|
August 6-11, 2006 11th International Congress of Pesticide Chemistry Kobe, Japan |
Dr.Hisashi Miyagawa |
|
August 27-31, 2006 European Chemistry Congress Budapest, Hungary |
Evelyn McEwan |
|
August 28 - September 2, 2006 6th International Symposium on Effects of Surface Heterogeneity in Adsorption and Catalysis on Solids (ISSHAC-6) Zakopane, Poland |
Wladek Rudzinski |
|
September 3-6, 2006 19th International Symposium on Chemical Reaction Engineering (ISCRE-19) Berlin, Germany |
Mrs. Sabine Sporleder |
|
September 3-6, 2006 New Industrial Limerick , Ireland |
Richard Holliday |
|
September 3-8, 2006 6th European Conference on Computational Chemistry High Tetras, Slovakia |
E-mail: cernusak@fns.uniba.sk |
|
September 7-12, 2006 12th International Conference on High Temperature Materials Chemistry Vienna, Austria |
Prof. Dr. Adolf Mikula |
|
September 10-14, 2006 9th International Symposium Louvain-la-Neuve, Belgium |
Marianne Saenen |
|
September 13-15, 2006 10th International Oxfordshire, UK |
Mrs. Carolyn Pickering |
|
October 2006 Asia Biofuels Conference Beijing, China |
Biofuelsconferences.com |
|
October 16-20, 2006 27th FLAQ International Congress on Chemistry Havana , Cuba |
Alberto Nunez |
|
October 16-20, 2006 27th Latinamerican Congress on Chemistry & Havana, Cuba |
Roberto Cao Vazquez |
|
October 18-20, 2006 12th Symposium on Sample Handling for Zaragoza , Spain |
Marianne Frei |
|
July 22-27, 2007 12th International Symposium on Novel Aromatic Compounds (ISNA-12) Tsuna-Gun, Japan |
Prof. Yoshito Tobe |
|
August 4-12, 2007 44th IUPAC General Assembly Turino, Italy |
IUPAC Secretariat |
|
August 5-11, 2007 41st IUPAC Congress "Chemistry Protecting Health, Natural Environment and Cultural Heritage" Turino, Italy |
IUPAC Secretariat |
|
September 7-14, 2007 EUROanalysisXIV Antwerp, Belgium |
E-mail: koen.janssens@ua.ac.be |

