
Organic Synthesis: Bifunctional catalyst weds reluctant partners
Matchmaking between ketones and olefins has never been easy. Although ketones readily pair up with many reagents, thanks to their ability to deprotonate and form nucleophilic enolates, olefins typically rebuff advances from such nucleophilic suitors. Now, chemists at the University of Texas, Austin, have found a catalyst capable of forging a carbon-carbon bond between this unlikely pair.
“Ketones are very common compounds. Olefins are even more common—they’re readily available feedstocks,” says Guangbin Dong, the chemistry professor who spearheaded the work. “We thought if we could find a good way to couple these two together, it would be very useful.”
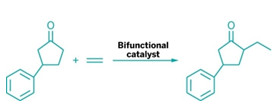 A bifunctional catalyst weds ketones to olefins
A bifunctional catalyst weds ketones to olefins
in a by-product-free strategy.
Dong and postdoc Fanyang Mo determined that the ideal matchmaker would be a bifunctional catalyst that contains both a secondary amine, 7-azaindoline, and a low-valent rhodium complex (Science 2014, DOI: 10.1126/science.1254465). The amine part of the catalyst activates the ketone by forming an enamine, which is amenable to oxidative addition by the rhodium part of the catalyst. The resulting rhodium hydride coordinates with the olefin, followed by migratory insertion and reductive elimination to generate an alkylated enamine. Hydrolysis produces an α-alkylated ketone and regenerates the catalyst.
Dong notes that the reaction has several advantages over traditional ketone alkylations, which typically are done by deprotonating the α-carbon of a ketone and using the resulting enolate to attack an alkyl halide. For example, he says, the new reaction tolerates a broad range of functional groups and is regioselective for alkylating the less hindered side of the ketone.
“This is a very cool piece of chemistry,” comments David W. C. MacMillan, an organic synthesis expert at Princeton University. “The capacity to use simple olefins as alkylating reagents for carbonyls is a transformation that is appreciated by the community to be valuable. Although a number of other groups have successfully demonstrated the use of olefins to perform the equivalent of enolate alkylation, this new variant is unique and could find broad application.”
Dong hopes to optimize the catalyst so that he might replace rhodium with a less expensive transition metal.
Nanotechnology: 92% of nanotubes generated with a new protocol have identical geometry, conductivity
Because of their strength, flexibility, and conductivity, carbon nanotubes might one day feature prominently in devices such as solar cells and miniaturized electronic circuits. Standard methods used to grow the tiny cylinders, however, typically produce a mixture of tubes with varying carbon-atom geometries and electrical properties, making their incorporation into gadgets problematic.
Now, Yan Li of China’s Peking University and coworkers have grown a batch of single-walled carbon nanotubes that are 92% pure (Nature 2014, DOI:10.1038/nature13434). The tubes in this pure portion have a single geometry—also known as chirality—so they behave identically: In this case, they have a uniformly high electrical conductivity, acting like metal wires.
To accomplish the high-purity growth, Li says her team found the “right recipe” for the nanocrystal catalysts that are used to seed nanotube growth. “People have long been thinking about using the structure of the catalyst to template the structure of nanotubes,” she says. But the best nanotube purity achieved previously was only 55%.
To seed the growth of highly uniform carbon nanotubes (gray), researchers use tungsten-cobalt alloy nanocrystals (yellow and orange), as shown in these side- and top-view illustrations.
 Her group succeeded where others failed, Li says, because it synthesized uniform tungsten-cobalt alloy nanocrystal catalysts that are stable at the high temperature needed to synthesize nanotubes. During the growth process, a silicon wafer decorated with nanocrystals gets heated to 1,030 °C. Ethanol flowing over the wafer vaporizes, its carbon atoms sticking to the nanocrystals and eventually forming cylindrical lattices.
Her group succeeded where others failed, Li says, because it synthesized uniform tungsten-cobalt alloy nanocrystal catalysts that are stable at the high temperature needed to synthesize nanotubes. During the growth process, a silicon wafer decorated with nanocrystals gets heated to 1,030 °C. Ethanol flowing over the wafer vaporizes, its carbon atoms sticking to the nanocrystals and eventually forming cylindrical lattices.
The way the tungsten and cobalt atoms pack together on the faces of the nanocrystal catalysts is essential for producing highly pure nanotubes, Li explains. Where the carbon atoms stick to the crystal facets dictates what nanotube geometry emerges.
“Li and coauthors have made a major breakthrough,” says Mohan Sankaran, a chemical engineer who studies nanotube growth at Case Western Reserve University. “Their approach will help realize the immense potential of carbon nanotubes.”
The researchers believe they can increase nanotube purity to above 99% by further manipulating the composition, structure, and size of their nanocrystal catalysts. Li says they’ve already adjusted their patented protocol to grow high-purity tubes of an entirely different geometry that behave like semiconductors.
Organic Chemistry: The one-pot reaction might simplify sustainable routes to fuels or fine chemicals from biomass
Researchers report a one-pot catalytic reaction to convert cellulose to n-hexane in high yield (ACS Sustainable Chem. Eng. 2014, DOI: 10.1021/sc5001463).
A direct path from cellulose to hexane has the potential to simplify routes for turning biomass into aromatic compounds like benzene or into isohexane, a component of gasoline, says Bert Sels of Catholic University of Leuven, in Belgium, who was not involved with the research. Such a process could effectively replace petroleum as a starting material for those chemicals.
Some scientists have worked on catalysts to break cellulose into molecules of glucose, which can then be transformed into other chemicals. However, converting cellulose to glucose and then to hexane requires multiple reactions, some of which may not produce enough for industrial applications. What’s more, some metal-based catalysts break up the cellulose into carbon chains smaller than hexane, meaning chemists would have to add in other reactions to rebuild the longer chain.
In 2013, Yoshinao Nakagawa and Keiichi Tomishige, together with their colleagues at Tohoku University, in Japan, used a catalyst to convert glucose directly to hexane (ChemSusChem, DOI: 10.1002/cssc.201200940). They wondered if this catalyst, made from iridium metal and rhenium oxide supported on silica, also could yield hexane directly from cellulose.
The researchers combined microcrystalline cellulose, their catalyst, and the zeolite HZSM-5 in water and heated the mixture to 210°C under an atmosphere of hydrogen gas. Acid in the pores of the zeolite hydrolyzes cellulose into glucose. Next, glucose and hydrogen react, with the help of the metal catalyst, to produce the six-carbon alcohol sorbitol. The zeolite and metal catalysts then convert sorbitol to hexane, which floats into a layer of dodecane above the water mixture, making it easy to recover the product, Nakagawa says. The overall process, starting from cellulose, produces hexane in 78% yield after 24 hours.
Typically, to help push the conversion of cellulose into other chemicals, researchers grind cellulose crystals into small pieces. The process enhances the reactivity of the cellulose but requires a lot of energy. So Atsushi Fukuoka of Hokkaido University, in Japan, is excited to see the high yields of hexane starting directly from microcrystalline cellulose, because that suggests it might be possible to eliminate the grinding step. The researchers, he says, next should test this new reaction using raw biomass instead of processed cellulose.
Bert M. Weckhuysen of Utrecht University, in the Netherlands, likes the idea of a one-pot process to convert cellulose to hexane but wonders about the lifetimes of the two catalysts. Also, he thinks it would be interesting to include a reaction that forms branched hydrocarbons for transportation fuels.
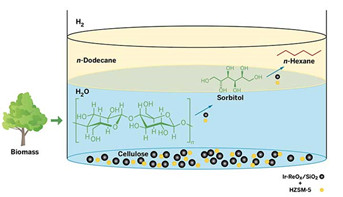
Cellulose Conversion
This one-pot process converts cellulose to sorbitol and then n-hexane in the presence of hydrogen gas. The multistep transformation involves two catalysts: an acidic zeolite HZSM-5 (yellow spheres), as well as iridium metal and rhenium oxide on silica support (black spheres). The reaction takes place in hot water, and the product, n-hexane, floats into a layer of n-dodecane solvent.
Organic Synthesis: Reaction eases addition of chiral carbon centers to aryl groups
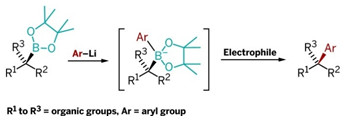
REVIVAL
This new reaction combines chiral secondary and tertiary boronic esters (left) with lithiated aryls to form intermediates that rearrange upon electrophile addition, yielding arylalkyl coupling products.
The revival of a nearly 50-year-old technique for forming carbon-carbon bonds may help ease the synthesis of aryl derivatives, such as drug candidates.
Varinder K. Aggarwal and coworkers at the University of Bristol, in England, have taken a venerable but neglected synthesis called Zweifel olefination and made it new again (Nat. Chem. 2014, DOI: 10.1038/nchem.1971).
The team’s approach overcomes key limitations of Suzuki-Miyaura coupling, the reaction most often used in the pharmaceutical industry for creating C–C linkages. In Suzuki-Miyaura coupling, a transition-metal catalyst is used to join sp2 carbon atoms (like those in aryl groups) to one another. It cannot easily link sp2 to sp3 (single-bonded) carbon atoms, especially in a stereocontrolled manner.
The new synthesis reported by Aggarwal and coworkers readily links sp2 carbon atoms to sp3 carbons with excellent stereocontrol and without use of transition metals. It reacts aryllithium reagents with chiral secondary or tertiary boronic esters to form aryl products with a chiral tertiary or quaternary carbon center (linked to three and four other carbons, respectively).
“It is a special paper,” says organoboron chemist Dennis G. Hall of the University of Alberta, Edmonton. “There is some precedent—there always is—but it achieves a feat that is virtually unachievable by regular Suzuki-Miyaura coupling.”
“It obviates difficulties introduced by some of the side reactions often seen in transition-metal chemistry,” comments another organoboron chemist, Cathleen Crudden of Queen’s University, in Kingston, Ontario. “The fact that it can be employed for the gener-ation of quaternary centers with high levels of enantiomeric purity is remarkable,” she says, noting that coupling reactions of that kind have generally not been available before.
Earlier, Aggarwal’s group developed an enantioselective way to make secondary and tertiary boronic esters. “We were keen to find a way of coupling them to aromatics but recognized that this was a very difficult problem using classical Suzuki-Miyaura-based conditions,” he says. “We wondered whether we could take a leaf out of the Zweifel olefination reaction reported almost 50 years ago.” That reaction uses vinyllithium to make vinyl-alkyl products (J. Am. Chem. Soc. 1967, DOI:10.1021/ja00990a061 and 1968, DOI: 10.1021/ja01024a068).
Aggarwal and coworkers substituted aryl for vinyl starting materials, optimized the process, and made it stereoselective. In the revived reaction, lithiated electronrich aromatics and heteroaromatics are added to chiral secondary and tertiary boronic esters to form boronate intermediates. An electrophile such as N-bromosuccinimide is then added to induce a stereospecific rear-rangement, eliminating the boronate and creating final products.
The new reaction does have drawbacks. It tolerates only a limited range of functional groups on the starting materials and requires electron-rich aromatics. Aggarwal and coworkers are currently trying to expand the reaction’s scope to a broader range of functional groups and aromatics.
Carbon Capture: The low-cost material can effectively absorb the greenhouse gas, even under wet and acidic conditions like those found in power-plant gas streams
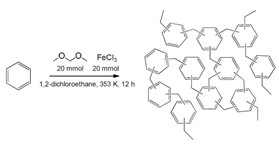
Knitted Benzene
To make a new carbon-capture polymer, chemists cross-linked benzene with formaldehyde dimethyl acetal using Friedel-Crafts alkylation.
An inexpensive material made of cross-linked benzene acts like a stiff sponge, swelling as it absorbs carbon dioxide. Unlike other carbon-capturing materials, it retains most of its absorbent ability under wet or acidic conditions (J. Am. Chem. Soc. 2014, DOI: 10.1021/ja5031968). The researchers think it could be used to collect CO
Some solid carbon-capturing materials are crystalline porous substances such as zeolites or metal-organic frameworks (MOFs). These rigid lattices have lots of pores to hold onto CO2, but they also can grab water, which is a common component of most gas streams at power plants. This means the materials capture less CO
Andrew I. Cooper of the University of Liverpool, in the U.K., and his colleagues wanted to find a substance that would target CO2 even with water in the mix. They developed a new polymer using Friedel-Crafts alkylation to cross-link benzene with formaldehyde dimethyl acetal. The resulting material looks and feels like a hard, brown chunk of plastic but is extremely porous.
Under dry conditions, the material could pick up 15.32 mmol of CO2 per gram, which is significantly more than other solid carbon-capture materials that they tested. Activated carbon, a polar zeolite, and two MOFs absorbed between 6.91 and 9.30 mmol/g. The cross-linked benzene sponge retained 86% of itsCO
The new material also is quite stable under acidic conditions like those found in power-plant gas streams. The team boiled the polymer in a sulfuric acid solution for an hour and found that its porosity and CO
The main disadvantage for the material is that its capacity surpasses the other carbon-capture materials only at high pressures. But most power plants vent CO2 at relatively low pressures, which don’t cause this material to swell. So for now, this particular polymer would be useful only in capturing carbon in high pressure gas streams such as those in plants that use integrated gasification combined cycle technology. These plants convert coal and other carbon-based fuels into a mixture of CO2 and H2 and then combust the H2 to produce energy. The precombustion gas streams have pressures high enough to allow the cross-linked polymer to swell. Unfortunately, such gasification plants are still rare, Cooper says.
Neil B. McKeown of the University of Edinburgh says that although the team has presented some methodical tests of the cross-linked polymer, there’s still a lot to prove before people will start to consider the material. Currently, he says, there’s competition between carbon-capture technologies, and “it’s not clear which is best, and none of them are great.”
But McKeown does say the polymer has one major advantage: It’s cheap to make. Cost counts, he says, when a material must be made in large enough quantities to capture the hundreds of tons of CO
Organic Synthesis: Process converts amines into nitrogen heterocycles of interest for drug discovery
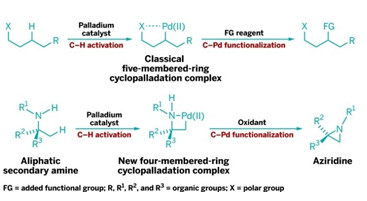
RING CYCLE
New C–H functionalization reaction proceeds via virtually unprecedented four-membered cyclopalladation intermediates (bottom row) instead of conventional five-membered (shown, top row) or six-membered ones.
Chemists have developed a unique way to get specific carbon-hydrogen bonds in organic compounds to break so new carbon-based connections can be made. The technique could make it easier to create a range of nitrogen-based compounds for drug discovery and other applications.
C–H bonds are notoriously unreactive, but in recent years researchers have developed ways to coax them into being more cooperative. Typically, this is done by cyclometalation, in which a metal such as palladium coordinates to a polar functional group and then breaks into an adjacent C–H bond to form a five- or six-membered cyclic intermediate. The carbon-metal bond in the cyclic intermediate can then react with a range of reagents, releasing the metal and forming products in which the C–H bond’s carbon is functionalized.
Matthew J. Gaunt and coworkers at the University of Cambridge have now discovered a family of catalytic reactions in which palladium coordinates with a methyl group adjacent to a secondary amine group, yielding a four-membered palladacycle intermediate (Nature 2014, DOI: 10.1038/nature13389). Such four-membered rings have been virtually unprecedented in cyclometalations.
Gaunt and coworkers found that the palladacycles can be transformed into strained nitrogen heterocycles such as aziridines and β-lactams by reacting them with reagents such as an oxidant. Gaunt believes the resulting products “will enable medicinal chemists to extend the range of chemical space accessible around the ubiquitous amine function.”
C–H functionalization specialist Gong Chen of Pennsylvania State University says the reaction is important because the unfunctionalized secondary alkyl amines it uses as starting materials have been underutilized for organic synthesis. Such amines are often used as bases or coupling partners but are seldom used to construct drug pharmacophores, he notes.
John F. Hartwig of the University of California, Berkeley, who specializes in catalytic reactions involving metal complexes, comments that “the application of C–H bond functionalization to the synthesis of amines, as opposed to sulfonamides or amides, is rare, and the ability to do so while forming a strained ring is particularly surprising.”
Gaunt’s group is currently working on expanding the technique into a more general strategy. For example, right now the new method is restricted to fairly hindered amines—secondary amine groups bearing bulky substituents. But preliminary results reported in the paper suggest that less hindered amines can undergo similar reactions. Gaunt and coworkers are trying to make the new process viable for all classes of secondary amines.
They would also like to better understand the mechanism and selectivity of the reaction, develop an enantioselective version, and apply it to targeted synthesis of important drugs and natural products, among other projects.
“This is a sweet discovery, highlighting the great potential of using C–H functionalization to develop new organic transformations,” Chen adds.
Catalysis: Study questions conventionally ascribed role of metal particles on semiconductors
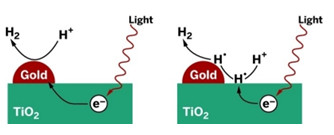
UNCONVENTIONAL
Metal particles on TiO2 are commonly regarded as electron-transfer sites active in production of H2 (left). A study suggests that the metals may be uninvolved in electron transfer (right).
Titanium dioxide’s knack for mediating sunlight-driven chemical reactions has made it the material of choice for a host of applications, including self-cleaning glass, antifogging coatings, and splitting water to make hydrogen. Researchers have known for years that modifying the semiconductor with metal nanoparticles increases its photocatalytic activity. But the conventional explanation for that enhancement has been called into question by a new study that challenges the mechanistic role of the metal nanoparticles.
The study may deepen understanding of photochemical processes and may lead to lower-cost catalysts.
Shining light on TiO2 or other semiconductors promotes electrons to an excited state from which they can stimulate chemical reactions. Much of the time, the electrons shed their excess energy far too quickly for reactions to commence. But if TiO2 is doped with platinum or other metal nanoparticles, excited electrons can quickly hop onto the metals and get trapped before they can cool down, according to the common explanation.
In the case of water splitting, that model indicates that the metals serve as tiny islands on the semiconductor surface on which hydrogen ions can congregate, undergo reduction with trapped electrons, and form H2.
A team led by University of California, Riverside, chemistry professor Francisco Zaera now proposes that electron transfer from the semiconductor to the metal may not play a significant role in photocatalysis. The team came to that conclusion by using transient-absorption and time-resolved-fluorescence spectroscopy methods to probe the photophysics and energy-transfer processes in a series of custom-made nanoparticles, including Au/TiO2 and Pt/TiO2 (Proc. Natl. Acad. Sci. USA 2014, DOI: 10.1073/pnas.1405365111).
The Riverside team proposes that in contrast to the electron-trap model, excited electrons stimulate H+ reduction on the surface of the semiconductor, not the metal. Hydrogen atoms then migrate from the semiconductor to the metal, which combines them catalytically to form H2.
Photochemistry: Compact fluorescent bulbs power production of desirable building blocks featuring delicate bonds because it uses visible light, the new method works even with substrates with weak C–Br bonds.

By combining two catalysts, researchers have used photochemis-try to build enantiomerically enriched rings in high yields. The advance could add to the tool kit for building motifs present in agrochemicals and pharmaceuticals.
For more than a century, chemists have talked about generating chirality with photochemistry because it can access products not available by other routes. But photochemistry’s march toward enantioselectivity has lagged behind those for transition-metal catalysis or organocatalysis. The reason is that once a molecule absorbs a photon of light, it reacts before it can be reined in by a stereochemistry-controlling catalyst, explains Tehshik P. Yoon, who led the new work. The few existing enantioselective options require specialized light sources or carefully designed catalysts.
Yoon and his coworkers at the University of Wisconsin, Madison, reported in Science a different approach. Their method makes chiral cyclobutanes from the visible-light-promoted [2 + 2] photocycloaddition of α,β-unsaturated ketones (2014, DOI:10.1126/science.1251511). It requires two catalysts: Ru(bpy)3, a transition-metal complex that absorbs visible light, and a chiral Lewis acid made from the lanthanide element europium.
“We’re using wavelengths of light that pass through organic molecules,” so they don’t enter the excited state that leads to willynilly reactivity, Yoon says. It’s Ru(bpy)3 that absorbs this light, which comes from a compact fluorescent bulb, and then “spits out an electron” to trigger cyclobutane ring formation, Yoon explains. The reaction occurs under chiral control because the chiral Lewis acid coordinates to the ketone substrate.
The work cleverly mimics photosynthesis in that it decouples the harvesting of light energy from bond-breaking and bond-forming steps, explains University of Neuchâtel, Switzerland, chemist Reinhard Neier in an accompanying commentary (DOI:10.1126/science.1252965).
“I think the concepts are general” and will yield more than just cyclobutanes, Yoon says.
Chemical & Engineering News
В январе 2015 г. Американское химическое общество (ACS Publications) планирует начать выпуск нового рецензируемого научного журнала ACS Central Science.
В этом междисциплинарном журнале будут публиковаться инновационные исследования, демонстрирующие основополагающую роль химии в различных областях науки. ACS Central Science предоставит ученым всего мира возможность сообщить о сделанных прорывных открытиях. Публикация в журнале будет бесплатной, опубликованные статьи будут находиться в открытом доступе.
Редколлегия нового журнала обеспечит высокое качество статей, своевременное рецензирование, быструю публикацию и широкое распространение информации.
Science is big. Growing rapidly. And increasingly interdisciplinary. Yet researchers remain immersed in their respective fields and sometimes miss the bigger, interconnected picture. ACS is about to change the way you look at science.
In early 2015, ACS Publications will launch a new peer-reviewed scientific journal unlike any other.
ACS Central Science will be much more than another chemistry journal—it will showcase exceptional, innovative research across the full spectrum of sciences that demonstrate the fundamental, transcending power of chemistry. It will give scientists around the world a prominent platform for showcasing their cutting-edge dis-coveries. And it will be open in every sense of the word: no publishing charges for authors and free access for all readers.
ACS Central Science will be led by an internationally acclaimed researcher whose vision will address the needs of the global scientific community within and beyond traditional chemistry. The journal will provide not just information, but inspiration for scientists across the entire breadth of the chemical sciences and related fields.






