

7 февраля исполнилось 60 лет заместителю директора Института органической химии им. Н.Д. Зелинского РАН, доктору химических наук, профессору Александру Юрьевичу Стахееву.
А.Ю. Стахеев – специалист в области синтеза и исследования структуры и свойств гетерогенных катализаторов процессов тонкого органического синтеза и защиты окружающей среды, автор и соавтор 120 научных работ и 5 патентов.
Одним из основных направлений работ Александра Юрьевича является разработка гетерогенных каталитических систем на основе моно- и биметаллических наночастиц, наночастиц интерметаллидов для процессов тонкого органического синтеза. Другое направление исследований связано с созданием новых каталитических методов охраны окружающей среды. Под руководством А.Ю. Стахеева разработаны высокоэффективные катализаторы удаления оксидов азота из выхлопных газов автотранспорта и тепловых электростанций, катализаторы удаления аммиака и летучих органических соединений из воздуха технологических помещений. В последние годы в руководимой им лаборатории катализа нанесенными металлами и их оксидами успешно развивается перспективное направление исследований, связанное с созданием композитных многофункциональных катализаторов и каталитических процессов для решения задач охраны окружающей среды.
А.Ю. Стахеев внес большой вклад в развитие фундаментальных и прикладных исследований в области каталитических превращений углеводородов на нанесенных наночастицах металлов и их оксидов, в создание научных основ приготовления катализаторов, содержащих в качестве активного компонента нанесенные металлические или оксидные наночастицы.
Александр Юрьевич читает курс лекций по гетерогенному катализу студентам 4-го курса Высшего химического колледжа РАН при РХТУ им. Д. И. Менделеева. Под его научным руководством защищены пять кандидатских диссертаций.
А.Ю. Стахеев – член диссертационных Советов Д 002.222.02 ИОХ РАН и Д 501.001.90, входит в состав экспертного совета ВАК, член редколлегий журналов “Кинетика и катализ”, “Катализ в промышленности” и “Успехи химии”, рецензент научных журналов “Известия АН. Серия химическая”, “Mendeleev Communications”, “Catalysis Communications”, “Catalysis Letters”, “Applied Catalysis B, Environmental”, “Catalysis Today”, “ASC Catalysis”, эксперт фонда РФФИ.
В 2016 году Российской академией наук А.Ю. Стахееву была присуждена премия имени А.А. Баландина за выдающиеся работы в области катализа (серия работ “Наноструктурирование активного компонента – метод управления каталитическими свойствами нанесенных металлических катализаторов в реакциях гидрирования и окисления”).
Научный совет по катализу ОХНМ РАН и редакция Каталитического бюллетеня сердечно поздравляют Александра Юрьевича с юбилеем и желают ему дальнейших творческих достижений!
Заседание Совета Международной ассоциации каталитических обществ
(International
Association of Catalytic Societies, IACS)
3 июля 2016 года в Пекине, непосредственно перед проведением 16-го Международного конгресса по катализу ICC-16, под председательством Президента IACS профессора А. Корма (Испания) состоялось заседание Совета Международной ассоциации каталитических обществ, под эгидой которого проводился Конгресс. На заседании Совета, кроме вопросов, связанных с проведением Конгресса, были рассмотрены следующие вопросы: принятие в Ассоциацию Румынского каталитического общества, определение места проведения 17-го Международного конгресса по катализу, выборы вице-президента Ассоциации, а также текущая работа Ассоциации (награды и почетное членство в IACS, обновление сайта Ассоциации).
16-й Международный конгресс по катализу
Профессор Кан Ли (Китай), председатель 16-го Международного конгресса по катализу, представил краткий обзор дел, связанных с проведением Конгресса. Работа Конгресса объединила более 2500 представителей из 50 стран. Около половины участников – граждане Китая. Конгресс был организован в виде 6 параллельных секций, тематика которых охватывает практически все направления науки о катализе. Более 300 выступлений, среди которых пять пленарных лекций, два выступления лауреатов премий Ассоциации (International Catalysis Award и Heinz Heinemann Award), 18 ключевых докладов, 81 приглашенный доклад и 201 устное сообщение. Всего было получено 2566 тезисов докладов. Представленные тезисы свидетельствуют о существенном повышении интереса исследователей к таким областям как конверсия биомассы, гидрирование CO2 и охарактеризование катализаторов в режиме in situ. По результатам оценки качества полученных тезисов присуждены 37 наград молодым ученым.
Голосование по вопросу членства Румынии в IACS
Румынское каталитическое общество впервые было зарегистрировано в 1965 году и перерегистрировано в 1992 году после произошедших в стране политических изменений. Общество объединяет 205 членов, работающих в университетах и исследовательских институтах. Участники заседания рассмотрели заявку, поданную Румынским каталитическим обществом, и единогласно ее одобрили путем тайного голосования. Профессор Корма поздравил нового члена Международной ассоциации каталитических обществ и пригласил представителей Румынии принять участие в дальнейшей работе заседания Совета IACS.
Место проведения 17-го Международного конгресса по катализу ICC-17
Выступили представители трех обществ – членов Ассоциации, которые претендовали на проведение следующего Международного конгресса по катализу в своих странах: профессор Михаэль Боукер от Великобритании, профессор Ганапати Ядав от Индии, профессор Энрике Иглесиа от Североамериканского каталитического общества. На тайном голосовании 22 голоса из 33 были отданы в пользу Североамериканского каталитического общества. В соответствии с результатами голосования профессор Корма объявил, что 17-й Международный конгресс по катализу будет проведен в Сан Диего (США) с 14 по 19 июня 2020 года. Тема Конгресса была определена как “Перспективы катализа в 2020 году”.
Новый состав Исполнительного комитета Ассоциации
В соответствии с Конституцией (Уставом) IACS, на период до следующего Международного конгресса по катализу в 2020 году Президентом IACS стал профессор Габриель Ченти (Италия), бывший до этого вице-президентом IACS. На пост следующего вице-президента IACS были выдвинуты два кандидата: профессор Энрике Иглесиа (США) и профессор Атсуши Фукуока (Япония). Выборы проходили путем тайного голосования без участия претендентов. Профессор Иглесиа был избран 24 голосами из 33. Были избраны также казначей, секретарь и другие члены Исполнительного комитета Ассоциации. В результате состав Исполнительного комитета до 2020 года стал следующим:
Президент – Габриель Ченти (Италия),
вице-президент – Энрике Иглесиа (США),
казначей – Джеймс Андерсон (Великобритания),
секретарь – Серджо Фуэнтес (Мексика),
управляющий делами – Атсуши Фукуока (Япония).
Обсуждение деятельности Ассоциации
Награды. В настоящее время Международная ассоциация каталитических обществ присуждает две награды: Международную премию по катализу и Премию Хайнца Хайнемана. Большинство участников заседания согласились с тем, что Премия Хайнца Хайнемана является престижной наградой. Она предназначена для присуждения не старейшим ученым – ветеранам науки о катализе, а тем, кто за достаточно сжатый период времени выполнил значительную работу, внесшую существенный вклад в развитие теоретического или прикладного катализа. Члены Совета обсудили периодичность присуждения этой премии. Большинством голосов был принят период в 10 лет (раньше это был промежуток между двумя Международными конгрессами по катализу, то есть четыре года). При этом было предложено учредить еще одну награду, за достижения в течение всей жизни. Это предложение также было поддержано большинством участников. Вопрос о новой награде будет проработан на следующем заседании IACS.
Почетное членство в IACS. Почетное членство в Международной ассоциации каталитических обществ получают специалисты, внесшие большой вклад в деятельность каталитического сообщества путем активного участия в работе и организации Международных конгрессов по катализу, отборе претендентов на получение наград Ассоциации и международном сотрудничестве в рамках IACS. Было решено, что Почетное членство присуждается по рекомендации каталитического сообщества не более чем трем претендентам на каждом Международном конгрессе по катализу.
Обновление сайта IACS. Сайт Международной ассоциации каталитических обществ появился 20 лет назад. Поскольку размещение новой информации на сайте связано со значительными сложностями, было признано необходимым создать новый сайт Ассоциации либо провести его существенное обновление на профессиональном уровне. Решением этого вопроса было предложено заняться молодым членам Ассоциации. В результате нынешним адресом сайта стал www.iacs-icc.org. Прорабатывается возможность создания общего сайта Международных конгрессов по катализу.
2-6 октября 2016 г., Светлогорск, Россия
http://conf.ict.nsc.ru/MCR-X/en
X Международная конференция “Механизмы каталитических реакций” была организована под эгидой Европейской федерации каталитических обществ (EFCATS) и Некоммерческого партнерства “Национальное каталитическое общество”. Конференция была проведена в бизнес-центре пансионата “Волна” г. Светлогорска со 2 по 6 октября 2016 при финансовой поддержке Российского фонда фундаментальных исследований и партнеров конференции: ООО “Реолгрейд сервис”, ООО “Фармконтракт Медиа”, SPECS Surface Nano Analysis GmbH, Frontier Laboratories Ltd., ООО “Катакон”.
На конференцию приехали 159 участников из 14 стран: были представлены Россия, Германия, Франция, Бельгия, Дания, Финляндия, Израиль, Италия, Япония, Румыния, ЮАР, Испания, Швейцария, США.
Организаторами конференции выступили:

Участники Конференции имели возможность прослушать и обсудить пленарные лекции, 7 ключевых лекций, 44 устных и 28 кратких устных докладов, а также 89 стендовых презентаций. В рамках Конференции была организована Молодежная школа-симпозиум по квантово-механическому моделированию – актуальному и востребованному способу изучения фундаментальных закономерностей каталитических реакций и процессов.

Работа Конференции проходила одновременно на трех устных сессиях. Тематика включала преимущественно вопросы, связанные с фундаментальными исследованиями закономерностей каталитических реакций, квантово-механического моделирования в катализе, дизайна новых каталитических систем, определения их физико-химических свойств и модификации в ходе проведения каталитических реакций; затрагивались вопросы разработки принципиально новых, а также модифицированных каталитических систем и процессов; особое внимание было уделено исследованию эволюции каталитических систем и реакционных сред в ходе проведения реакций методами in situ и operando, а также теории и моделированию в катализе:
Пленарные лекции затрагивали актуальные вопросы, связанные с возможностями катализа в решении энергетической проблемы, в том числе с использованием возобновляемого сырья, рециклом галогенсодержащих веществ и развитием современных способов изучения каталитических систем.
 Научную сессию открыла пленарная лекция лауреата международной Премии “Глобальная энергия” 2016 года академика РАН, проф.
Научную сессию открыла пленарная лекция лауреата международной Премии “Глобальная энергия” 2016 года академика РАН, проф.
Лекция д-ра С. Хелвега (S. Helveg), представленная д.х.н. А.Л. Кустовым (оба – Haldor Topsøe), затрагивала актуальные аспекты использования электронной микроскопии в каталитических исследованиях. К настоящему моменту, благодаря повышению чувствительности и разрешения, а также разработке газовых ячеек, стало возможно использовать электронную микроскопию для in situ исследования состояния гетерогенных катализаторов в ходе протекания каталитической реакции при давлениях вплоть до атмосферного. В сочетании с исследованиями каталитических свойств, электронная микроскопия открывает возможность “живого” наблюдения за строением/свойствами работающего катализатора на атомном уровне.
Галогены играют значимую роль в производстве ряда ценных химических соединений: полимеров, фармпрепаратов, поликарбонатов, и могут использоваться для селективной функционализации С-Н связей легких алканов в мягких условиях. Однако применение галогенов в химическом производстве требует создания замкнутых циклов их переработки. Лекция проф. Х. Переса-Рамиреса (J. Pérez-Ramírez, Institute for Chemical and Bioengineering, ETH Zurich, Switzerland) представляла собой обзор исследований по разработке катализаторов рециклизации галогенов (посредством окисления/оксигалогенирования) за последние десятилетия. Основной акцент был сделан на установлении взаимосвязей между синтезом, строением и свойствами гетерогенных катализаторов переработки галогенов.
В лекции проф. Д.Г. Влахоса (D.G. Vlachos, University of Delaware, Newark, USA) рассматривались фундаментальные проблемы, связанные с разработкой каталитических процессов и технологий получения топлив и химических продуктов из лигноцеллюлозной биомассы в качестве возобновляемого сырья. Докладчик продемонстрировал возможности применения ряда методов (кинетических, изотопных, спектроскопических и математических) для изучения механизмов каталитических процессов переработки возобновляемого сырья. В качестве конкретных примеров описано изучение механизмов реакций Дильса-Альдера и дегидрирования, применяемых в производстве п-ксилола, а также реакции гидродеоксигенирования, которая дает возможность осуществить восстановление фурана с целью получения топливных добавок и предшественников ароматических соединений.
В ключевой лекции проф. А. Кноп-Герике (A. Knop-Gericke, Fritz-Haber Institute of the Max-Planck Society, Berlin, Germany) был представлен ряд новаторских подходов, позволяющих исследовать поверхности жидкость-твердое тело с помощью методов in situ рентгеновской фотоэлектронной спектроскопии (РФЭС). Так, разработан подход к изучению методом РФЭС наночастиц, нанесенных на графеновую мембрану. Обсуждались возможности и ограничения данных подходов; особое внимание уделено дизайну новых исследовательских установок и ячеек.
Лекция д-ра Е. Кондратенко (E. Kondratenko, Leibniz Institute for Catalysis, Rostock Germany) была посвящена исследованию катализаторов метатезиса этилена и 2-бутена в пропилен полимеризационной чистоты на основе оксидов молибдена, нанесенных на Al2O3-SiO2. Выявлена зависимость между строением поверхностных металл-оксидных частиц и каталитической активностью: по сравнению с изолированными, полимерные MeOx структуры обеспечивают более высокую селективность за счет подавления изомеризации 2-бутена в 1-бутен.
Проф. Р. Лейно (R. Leino, Abo Akademi University, Turku, Finland) сделал обзорный доклад по использованию комбинации каталитических и кинетических методов для синтеза малых молекул. Подчеркнута современная тенденция к комбинированию методов органического синтеза, биокаталитических методов, а также гетерогенно- и гомогенно-каталитических методов (с использованием металлокомплексных и органокатализаторов). Обсуждались перспективы данного направления исследований.
Д-р И.В. Юданов (Институт катализа СО РАН, Новосибирск) рассказал о теоретических исследованиях с использованием квантовохимического моделирования размерного эффекта для адсорбции СО на поверхности нанокластеров платины и рассмотрел влияние сжатия решетки на адсорбцию водорода на поверхности наночастиц палладия и платины. Полученные данные интересны не только с фундаментальной, но и с практической точки зрения, поскольку позволяют разработать подходы к управлению каталитическими свойствами за счет варьирования размеров наночастиц катализатора.
Д-р А.Б. Сорокин (A.B. Sorokin, Institut de Recherche sur la Catalyse et l'Environnement de Lyon, France) рассказал о получении, строении и каталитических свойствах высоковалентных биядерных комплексов железа, стабилизированных макроциклическими (фталоцианиновыми и порфириновыми) лигандами. Полученный в ходе работы биядерный биомиметический комплекс [LFeIV-N=FeIV(L+•)] с азотным мостиком охарактеризован набором физических методов и оказался высокоактивным в ряде процессов активации С-X связей, в частности прямого окисления метана и окислительных превращений ароматических С-F групп в чрезвычайно мягких условиях. Представлен вероятный механизм каталитического действия описанного комплекса.
Проф. И.В. Коптюг (Международный томографический центр, Новосибирск) представил результаты исследования механизма ряда каталитических превращений методом ЯМР с использованием гиперполяризованного параводорода. В частности, данный метод позволил зафиксировать интермедиаты и изучить механизм процессов гидрирования ненасыщенных углеводородов на металлокомплексных катализаторах в растворе. Показано, что разработанный подход может быть успешно распространен на реакции гидрирования на гетерогенных катализаторах, включая иммобилизованные металлокомплексы, нанесенные и ненанесенные металлы и оксиды металлов. Другим перспективным способом изучения механизмов каталитических реакций является использование в ЯМР-исследованиях спиновых изомеров симметричных молекул (в частности этилена).
В лекции проф. С.А. Шаурман (S.A. Schauermann, Institute of Physical Chemistry, Kiel, Germany) представлены исследования механизма ряда сложных гетерогенно-каталитических процессов с помощью комбинации молекулярно-пучковых, спектроскопических и калориметрических методов в условиях высокого вакуума. Изучен механизм реакции селективного гидрирования акролеина в ненасыщенный спирт на Pt(111). Установлены особенности механизма хемосорбции H2O на поверхности Fe3O4(111)/Pt(111), протекающего через стадию быстрой диссоциации H2O с образованием димероподобного гидроксил-водного комплекса. Эти исследования опровергают общепринятый механизм диссоциации воды, включающий образование двух индивидуальных ОН-групп.
В ключевой лекции, представленной на Молодежной школе-симпозиуме по квантово-механическому моделированию, проф. С. Фабрис (S. Fabris, CNR-IOM DEMOCRITOS, Trieste, Italy) рассказал о применении комбинированных теоретических методов (DFT в сочетании с неэмпирическими методами молекулярной динамики, метадинамики и др.) для моделирования поверхности платиновых электродов. Были также представлены разработанные теоретические подходы к оценке свойств электродов в разных условиях: варьирующихся от вакуума при абсолютном нуле температур до реальных электродов в растворах при комнатной температуре.
В ключевой лекции проф. К.М. Неймана (K.M. Neyman, ICREA & University of Barcelona, Barcelona, Spain) был описан новый теоретический подход по установлению атомного упорядочения в наносплавах, пригодный для описания как наносплавов переходных металлов, так и наносплавов переходных металлов с s,p-элементами. Метод позволяет предсказывать стабильность трехмерной структуры наносплавов, прослеживать структурные изменения, происходящие при повышении температуры, взаимодействии с атмосферой и подложкой. Метод может применяться на практике для инжиниринга нестандартных наносплавов.
Таким образом, ключевые лекции продемонстрировали возможности новых усовершенствованных методических подходов в изучении фундаментальных закономерностей протекания важных каталитических процессов, обеспечивающих подходы для создания новых каталитических систем с перспективными свойствами. Особенно следует отметить, что не только за рубежом (Германия, Франция, Финляндия и др.), но и в России (например, в ИК СО РАН) успешно развиваются перспективные и новаторские подходы к исследованию катализаторов и каталитических процессов.
Программу устных докладов Секции I открыла д-р Л. Москалева (L. Moskaleva, University of Bremen, Bremen, Germany). Ее доклад был освящен проблеме компьютерного моделирования процессов, происходящих на поверхности кластеров нанопористого золота. Доклад д-ра Д.А. Пичугиной (Московский государственный университет имени М.В. Ломоносова) также был посвящен теоретическому изучению процессов, происходящих на малых кластерах золота. Было проведено сравнение активности кластеров золота со сходными по строению кластерами палладия и серебра. В работе к.х.н. Е.А. Шор (Институт химии и химической технологии СО РАН, Красноярск) с помощью теории функционала плотности (DFT) исследовано формирование активных центров реакций окисления в катализаторах на основе оксида церия и кластеров серебра. К.х.н. И.Ю. Скобелев (Институт катализа СО РАН, Новосибирск) рассказал об исследовании механизма окисления органических веществ перекисью водорода на титансодержащих полиоксометаллатах методом функционала плотности. Работа к.х.н. А.В. Матвеева (Новосибирский государственный университет) была посвящена поиску корреляции между структурой и каталитической активностью кластеров металлов – Pt, Rh, Ir, Fe, Re и Ru. К.х.н. Т.В. Тюмкина (Институт нефтехимии и катализа, Уфа) в своем докладе затронула проблемы моделирования механизма трансформации металлоциклических соединений. К.х.н. Е.А. Лашина (Институт катализа СО РАН, Новосибирск) рассказала об эффекте гистерезиса и автоколебаниях в окислении монооксида углерода на палладиевой фольге.
Секция II была посвящена современным методам изучения механизмов каталитических реакций. В докладах были представлены новые способы исследования механизмов с использованием современных физико-химических методов, включая методы in situ, а также новые способы обработки данных, открывающие перспективные возможности для анализа поверхностных структур. В этом смысле тематика секций II и III оказалась пересекающейся. Так, к.х.н. Е.В. Голубина (Московский государственный университет имени М.В. Ломоносова) представила интересный доклад об изучении методами in situ и ex situ (РФЭС, EXAFS, ТПВ и т.д.) процессов формирования активного компонента катализаторов на основе наноалмазов с нанесенным никелем. В докладе показано, что не слишком распространенный способ обработки результатов EXAFS исследования с применением вейвлет-преобразования дает возможность различить атомы легких элементов (например, кислорода и углерода) в локальном окружении каталитически активного атома (в данном случае никеля). Проф. М. Нагаи (M. Nagai, Tokyo University of Agriculture and Technology, Koganei, Japan) в своем докладе затронул тему изучения активных центров углеродных материалов – катализаторов электрохимического восстановления кислорода в топливных элементах и реакции Фриделя-Крафтса.
Следующие три секционных доклада представили сотрудники Института катализа СО РАН (Новосибирск). Д.х.н. В.А. Садыков рассказал о результатах изучения механизма риформинга этанола в синтез-газ на оксидных катализаторах, допированных платиной, никелем и рутением, методом SSITKA в комбинации с калориметрией и ИК-спектроскопией in situ. К.х.н. О.А. Стонкус – о применении методов просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) и рентгеновской фотоэлектронной спектроскопии для изучения механизма окисления монооксида углерода. К.х.н. В.Л. Кузнецов – об исследованиях особенностей формирования углеродных нанотрубок с использованием комплекса современных физико-химических методов, включая in situ РФЭС и ex situ ПЭМВР.
Доклад к.х.н. В.В. Живонитко (Международный томографический центр, Новосибирск) был посвящен возможностям применения параводорода для изучения механизмов каталитических реакций методом ЯМР. Исследованию механизма метатезиса олефинов был посвящен и доклад к.х.н. М.Л. Грингольц (Институт нефтехимического синтеза РАН, Москва). В этой работе метатезис проводили на катализаторах Граббса 1-го и 2-го поколения. К.х.н. Г.А. Бухтиярова (Институт катализа СО РАН, Новосибирск) представила результаты изучения механизмов процесса гидродеоксигенации кислородсодержащих компонентов биомассы на катализаторах на основе силикагеля, оксида алюминия и фосфида никеля. Темой доклада д.х.н. С.Л. Иоффе (Институт органической химии РАН, Москва) было рассмотрение новых путей реакционной активности алифатических азотсодержащих органических веществ. К.х.н. Е.А. Козлова (Институт катализа СО РАН, Новосибирск) посвятила свой доклад изучению механизмов трансформации сульфидных фотокатализаторов, допированных благородными металлами, в реакции фотокаталитического выделения водорода из водных растворов неорганических доноров электронов. К.х.н. О.Т. Касайкина (Институт химической физики СО РАН, Москва) рассказала об исследовании влияния магнитного поля на каталитическое разложение перекиси водорода в системе, содержащей мицеллы.
На секции III особое внимание уделялось in situ и operando исследованиям механизмов гетерогенных каталитических реакций. К.х.н. М.Ю. Смирнов (Институт катализа СО РАН, Новосибирск) представил результаты исследования активности различных форм кислорода на поверхности платины. Профессор
Значительная часть докладов Секции IV также включала использование методов in situ. В частности, к.х.н. Т.Ю. Кардаш (Институт катализа СО РАН, Новосибирск) рассказала об исследовании окислительно-восстановительных трансформаций систем на основе оксида меди методом рентгенофазового анализа in situ. На секции присутствовали доклады об изучении фундаментальных основ процессов переработки биомассы. Например, доклад проф. О.П. Таран (Институт катализа СО РАН, Новосибирск), посвященный кинетическим исследованиям механизма превращения целлюлозы в глюкозу. Большой интерес вызвал доклад д-ра М. Розе (M. Rose, RWTH Aachen University, Germany) о кинетическом и механистическом изучении гетерогенного каталитического жидкофазного аминирования и изомеризации биогенных спиртов.
В ряде выступлений были рассмотрены механизмы окислительных процессов. Д.х.н. М.Ю. Синев (Институт химической физики РАН, Москва) рассказал об исследовании механизма окислительного сочетания метана на смешанных катализаторах NaWMn/SiO2, д-р А. Хенкин (A. Khenkin, Weizmann Institute of Science, Rehovot, Israel) – о механизме окисления С-Н связи на гомогенных катализаторах на основе полимолибдата.
На конференции были представлены результаты исследования механизмов многих других востребованных каталитических процессов. К.х.н. Р.В. Оттенбахер (Институт катализа СО РАН, Новосибирск) рассмотрел механизм асимметрического эпоксидирования олефинов в присутствии биомиметических комплексов марганца с использованием различных окислителей. Д-р А. Тирсоага (A. Tirsoaga, University of Bucharest) сделала сообщение об изучении дезактивации нанесенных катализаторов на основе оксида меди в реакции образования C-N связи между анилином и бромбензолом. Особый интерес слушателей вызвал доклад проф. Г. Кольба (G. Kolb, Fraunhofer ICT-IMM Bereichsleiter Energie- und Chemietechnik, Mainz, Germany) о детальном изучении механизма парового риформинга метанола на катализаторах Pt/In2O3/Al2O3 в микроканальных реакторах. Д-р
В докладах Секции V были представлены исследования самого широкого круга каталитических процессов, включая окисление, восстановление, изомеризацию, гидроочистку нефтяных продуктов, а также превращений компонентов, полученных из биомассы. Открывал секцию доклад к.х.н. О.Б. Бельской (Институт проблем переработки углеводородов СО РАН, Омск) об изучении путей трансформации тринитробензойной кислоты и создании катализаторов этого процесса. Д-р И. Делидович (I. Delidovich, RWTH Aachen University, Aachen, Germany) рассказала о механизме катализируемой фосфатами изомеризации альдоз в кетозы. Д.х.н.
Приятно отметить, что на конференции были широко представлены работы молодых ученых из России и зарубежных стран, причем уровень многих докладов не уступал докладам более маститых коллег. Порадовал и тот факт, что во многих далеких от столицы исследовательских центрах имеется современное научное оборудование для физико-химических исследований, позволяющее проводить детальное изучение каталитических реакций и их механизмов и публиковать данные в журналах с высокими импакт-факторами. Организаторы стремились предоставить возможность устного выступления максимальному числу молодых участников конференции, поэтому уже в первый день работы стартовала секция коротких (10 мин.) устных презентаций. Прекрасный доклад С.А. Черняка (Московский государственный университет имени М.В. Ломоносова) был посвящен гидрированию СО и диспропорционированию СО на частицах кобальта, нанесенных на нефункционализированные и функционализированные органическими группами нанотрубки. К.ф.-м.н. С.С. Якушкин (Институт катализа СО РАН, Новосибирск) представил результаты исследования гидрирования нитробензола на поверхности катализаторов Au/Al2O3 методом ЭПР. К.х.н. Л.С. Кибис из того же института рассказала об исследовании состояния частиц родия в реакции окисления СО. К.В. Семикин (Санкт-Петербургский государственный университет) представил результаты исследования гетерогенного алкилирования бутана. М.А. Салаев (Томский государственный университет) рассказал о природе синергетического эффекта в реакции окисления спиртов на серебряных катализаторах. А.В. Воротынцев (Нижегородский государственный университет) представил результаты исследования каталитического восстановления SiCl4 и GeCl4. В.В. Гриненко (Институт органической химии РАН, Москва) рассказал о механизме электрохимического преобразования ароматических соединений. К.х.н. А.М. Зима (Институт катализа СО РАН, Новосибирск) представила результаты исследования электронной структуры активных центров оксофериловых катализаторов. В.Ю. Евтушок (Институт катализа СО РАН, Новосибирск) рассказал об исследованиях механизма селективного окисления углеводородов перекисью водорода. В.Ю. Торбина (Томский государственный университет) рассказала об окислении пропиленгликоля пероксидом водорода. Ю.А. Чумаченко (Институт проблем переработки углеводородов СО РАН, Омск) представила результаты исследования эффекта влияния платиновых прекурсоров на формирование активных центров бифункциональных катализаторов гидрокрекинга биомасел. С.О. Стрекалова (Институт органической химии РАН, Москва) рассказала о процессах каталитической трансформации ароматических углеводородов.
Высокий уровень исследований продемонстрировали молодые ученые на молодежной секции, посвященной in situ исследованиям. Крайне интересные in situ исследования представили молодые ученые из Южного федерального университета Ростова-на-Дону. Так, к.ф.-м.н. К.А. Ломаченко рассказал о результатах изучения состояния меди в условиях реакции восстановления окислов азота методом спектроскопии края рентгеновского поглощения, а к.ф.-м.н. А.Л. Бугаев представил данные исследования формирования гидрида и карбида палладия в условиях каталитических реакций. Таким образом, в Ростове-на-Дону уже работает новый очень хорошо оснащенный научный центр каталитических исследований, в котором каталитические разработки ведутся на мировом уровне.
Но и известные каталитические центры “с историей”, например, Институт катализа им. Г.К. Борескова СО РАН, не сдают позиций. Так, на данной секции были представлены интересные доклады молодых ученых этого института: к.х.н. О.А. Булавченко провела фундаментальное исследование процесса восстановления оксидов на основе марганца и циркония методами рентгеновской дифракции и РФЭС. К.х.н. Д.И. Потемкин сообщил о кинетических исследованиях низкотемпературного риформинга легких углеводородов в присутствии метана на никелевых катализаторах. К.х.н. А.В. Нартова представила результаты исследования процесса дезактивации платиновых катализаторов при восстановлении окислов азота. К.ф.-м.н. А.А. Сараев применил метод атомно-слоевого осаждения для создания модельных катализаторов V2O5/TiO2(110).
К.х.н. В.В. Дутов (Томский государственный университет) провел in situ исследование окисления СО и этанола на поверхности катализаторов Ag/SiO2. Вторая реакция представляет значительный практический интерес. Похожие системы исследовал к.х.н. О.Г. Сальников (Международный томографический центр СО РАН, Новосибирск), используя в работе методы ядерного магнитного резонанса для изучения механизмов реакций гидрирования карбонильных соединений.
В программу Конференции была включена молодежная школа-симпозиум “Квантово-математическое моделирование каталитических процессов”. Широкое участие в конференции молодых ученых позволило им напрямую пообщаться с активно работающими профессорами российских и зарубежных ВУЗов и научных организаций, перенять опыт представления докладов, завязать новые научные контакты с коллегами.
В докладе к.х.н. С.С. Лалетиной (Институт химии и химической технологии СО РАН, Красноярск) были представлены результаты квантово-химических расчетов дегидрирования метанола на поверхности платиновых кластеров. В МГУ имени М.В. Ломоносова активно ведутся исследования катализа с участием золотосодержащих катализаторов расчетными методами. Так, две аспирантки университета – М. Голосная и Н. Никитина – рассказали о результатах квантовохимических расчетов структуры кластеров золота, причем во втором докладе основной задачей было определение состава каталитически активных центров. В докладе Ю. Полынской, также из МГУ имени М.В. Ломоносова, изучено каталитическое действие биметаллических золотосодержащих кластеров в процессе эпоксидирования пропилена на поверхности AgAu катализаторов. К.х.н С.Е. Малыхин (Институт катализа СО РАН, Новосибирск) представил результаты расчетов механизма реакции окисления олефинов окислами азота. Г.Ю. Жигулин (Институт металлоорганической химии РАН, Москва) проанализировал результаты расчетов эффекта влияния растворителя на образование комплексов меди. К.х.н. А.В. Матвеев (Институт катализа СО РАН, Новосибирск) рассказал о моделировании адсорбции кислорода на поверхности палладия.

Стендовая секция конференции прошла в атмосфере творческих дискуссий и позволила всем участникам пообщаться и обменяться знаниями и подходами, молодым ученым – получить консультации и советы более опытных коллег, более опытным – присмотреть среди аспирантов и студентов перспективных молодых исследователей для своих научных групп.

Значительную помощь в проведении конференции оказал Балтийский федеральный университет им. И. Канта. Принято решение о развитии сотрудничества между Институтом катализа СО РАН и БФУ им. И. Канта, в том числе о поддержке академической мобильности сотрудников этих организаций по проблемам исследования углеродных наноматериалов и нанотехнологий.
Организационный комитет наградил почетными Дипломами и премиями трех молодых участников, представивших новаторские исследования с использованием современных подходов и получивших результаты высокого уровня. Среди них два представителя России и один участник из Германии. Дипломы получили Маркус Розе (Marcus Rose, Institut für Technische und Makromolekulare Chemie, RWTH Aachen University, Aachen, Germany), Кирилл Ломаченко (Южный федеральный университет, Ростов-на-Дону) и Кирилл Семикин (Санкт-Петербургский технологический университет, Санкт-Петербург).
Сборник тезисов конференции выпущен на электронном носителе. Материалы устных сообщений планируется опубликовать в специальном томе журнала Кинетика и катализ.
Тематика Х Конференции отразила все тенденции, наблюдающиеся в современных исследованиях по катализу в мире: рост числа работ, связанных с переработкой возобновляемых ресурсов (природные соединения, биотоплива), фотокатализом, изучением наноструктурированных катализаторов, в том числе содержащих различные типы наноуглерода; учетом воздействия реакционной среды на катализатор.
Следует отметить, что конференция “Механизмы каталитических реакций” изначально создавалась в 1974 году как российская, но затем в 2009 году вышла на международный уровень. Конференция проводилась в Москве (1974, 1979, 1986, 1990, 2002 гг.), Новосибирске (1982, 2009 гг.), Санкт-Петербурге (2006, 2012 гг.) и Светлогорске (2016 г.). Международные МКР-конференции проходят под эгидой Европейской ассоциации каталитических обществ (EFCATS), НП “Национальное каталитическое общество” и посвящены высоко востребованной в мировом масштабе проблематике. Если раньше значительную часть иностранных участников составляли представители учебных и научных учреждений России, уехавшие работать за рубеж, то теперь растет число иностранных специалистов, привлеченных высоким научным уровнем конференции. К сожалению, уменьшается количество участников из стран СНГ (Беларуси, Азербайджана, Узбекистана и др.), что было отмечено и на последнем Международном конгрессе по катализу (2016 г., Пекин, Китай). Следует признать, что данный факт отражает объективно существующие в научной сфере СНГ тенденции.

Однако, вопреки насаждаемым в последнее время в общественных дискуссиях представлениям о невысоком уровне российской науки, X Международная конференция “Механизмы каталитических реакций” продемонстрировала, что в области каталитической химии работа идет на высоком научном уровне. В том числе и исследования многих молодых ученых, которые выгодно отличаются новаторскими подходами, широким использованием современных методов, творческим анализом полученных результатов.
По сравнению с предыдущими конференциями, более широко были представлены экспериментальные методы исследования механизмов каталитических реакций in situ и operando в сочетании с квантовохимическими методами анализа возможных интермедиатов, маршрутов их трансформации и структуры активных центров. Практически на всех, а не только на специализированных секциях, преобладали доклады о работах, выполненных с использованием этих подходов. Большое внимание было уделено результатам, полученным с помощью источников синхротронного излучения в международных центрах, как в России (Сибирский центр синхротронного и терагерцового излучения, Новосибирск), так и за рубежом (Берлинский источник синхротронного излучения BESSY-II, Германия). Прогресс в развитии фотоэлектронной спектроскопии позволил исследовать не только поверхность раздела твердое тело – газ, но и твердое тело – жидкость, что хорошо согласуется с международными тенденциями исследований в области катализа.

Также заметно, что на Х Конференции были представлены работы по самым разным направлениям каталитической науки, без преобладания реакций определенного типа, как это случалось на предыдущих конференциях. Это говорит о разнообразии запросов промышленности в отношении разработки научных основ каталитических процессов.
Важно подчеркнуть, что современное передовое научное оборудование дорого для закупки и в эксплуатации, поэтому координация работ между каталитическими научными центрами в мировом масштабе очень важна для развития всей науки о катализе. Проведение конференции, несомненно, будет способствовать развитию международного сотрудничества; как показали представленные на конференции доклады, в России имеются группы и центры, проводящие каталитические исследования на хорошем международном уровне.

На церемонии закрытия был отмечен высокий уровень организации конференции, творческая, деловая и дружеская атмосфера, созданная организаторами и участниками. Для проведения следующей XI Международной конференции “Механизмы каталитических реакций” в 2018 году выбран один из крупнейших экономических, культурных и научных центров России – г. Казань.
Материал подготовили В.И. Бухтияров, К.П. Брыляков,
В.В. Каичев, Е.А. Козлова, В.А. Садыков, Л.Я. Старцева
(ИК СО РАН, Новосибирск); Е.С. Локтева (МГУ, Москва)
Профессор, Заслуженный химик России
Евгений Голосман

Недавно в столице Урала Екатеринбурге прошел XX Менделеевский съезд по общей и прикладной химии. В один из дней работы съезда состоялся и отчетно-выборный съезд Российского химического общества имени Д.И. Менделеева.
Озвучил приветствие съезду президента РФ В.В. Путина полномочный представитель главы государства в Уральском округе Игорь Холманский. Съезд приветствовал губернатор Свердловской области Евгений Куйвашев. Руководитель федерального агентства научных организаций Михаил Котюков поздравил съезд от имени председателя Правительства РФ Д.А. Медведева. Вице-президент РАН академик Сергей Алдошин выразил уверенность, что съезд позволит определить основные пути развития отрасли на ближайшие пять лет. Председатель Уральского отделения РАН академик Валерий Чарушин на съезде и при открытии выставки напомнил девиз заводчиков Демидовых – “делами, а не словами”. Съезд приветствовала президент Международного союза по теоретической и прикладной химии (ИЮПАК) чл.-корр. РАН Наталья Тарасова. Приветствовали съезд по видеосвязи бессменный президент многих Менделеевских съездов академик Олег Нефедов и председатель Российского фонда фундаментальных исследований академик Владимир Панченко. Президент Российского химического общества академик Аслан Цивадзе напомнил, что еще недавно Российские города, кроме Москвы и Санкт-Петербурга, не были готовы к приему многотысячного съезда. Столица Урала сумела организовать столь грандиозное мероприятие.
Организаторами съезда выступили Российская академия наук, Уральское отделение Российской академии наук, Федеральное Агентство научных организаций, Правительство Свердловской области, Российское химическое общество имени Д.И. Менделеева, Министерство образования и науки Российской Федерации, Уральский федеральный университет имени первого президента России Б.Н. Ельцина, Национальный комитет российских химиков, Российский союз химиков. Съезд проводился под эгидой Международного союза по теоретической и прикладной химии (IUPAC).
Первый Менделеевский съезд памяти Д.И. Менделеева “Вопросы общей химии, отраслей химической технологии и проблемы приложения химии в разных отраслях науки и техники” состоялся в Санкт-Петербурге в 1907 г. В работе съезда приняли участие представители 80 городов. Позже съезды проводились в Москве, Ленинграде (Санкт-Петербурге), Казани, Харькове, Киеве, Алма-Ате, Баку, Ташкенте, Минске, Волгограде.

Группа участников I Менделеевского съезда
Тематика съездов охватывает основные направления развития химической науки, технологии и промышленности, химического образования и взаимодействия бизнеса с наукой и промышленностью, что существенно отличает их от обычных тематических научных конференций. Менделеевские съезды организуются с интервалом в четыре – пять лет в крупнейших научных центрах страны, и определяют пути развития химической науки и промышленности России.
В крупнейшем Российском научном форуме в Екатеринбурге приняли участие около 2000 делегатов, в том числе 300 иностранных ученых, а также представители компаний, занимающихся производством химических продуктов и материалов.
Среди 2000 участников ХХ съезда Нобелевские лауреаты, более 100 академиков и членов-корреспондентов Российской академии наук, несколько сотен профессоров и кандидатов наук, 700 молодых ученых. В работе съезда так же приняли участие: президент Российской инженерной академии чл.-корр. РАН, лауреат премии Правительства РФ Б.В. Гусев и другие действительные члены этой академии; академик, лауреат Государственной премии и премии “Энергия” В.Н. Пармон; академик РАН, Заслуженный нефтехимик СССР, лауреат Премии Правительства РФ С.Н. Хаджиев; лауреат премии Правительства академик В.В. Лунин; академик, лауреат Государственной премии М.П. Егоров и др. С устными и стендовыми докладами выступили более 1100 ученых. В 5 томах материалов съезда опубликовано около 2500 тезисов докладов от имени 7800 авторов на русском и английском языках.
Обсуждение многих химических проблем, и в том числе химического образования и преподавания в РФ, взаимодействия химической науки и бизнеса, актуальных проблем азотной промышленности, помимо пленарных, устных и стендовых докладов, проводилось и в рамках 9 секций, 3 Международных симпозиумов, 10 круглых столов, которые представляют великолепную площадку для дискуссий специалистов науки и промышленности, определяющих направление и развитие металлургической, нефтехимической и других отраслей промышленности.
Конечно же, осуществление столь масштабного мероприятия было бы затруднительным без финансового вклада спонсоров и десятков организаций. Большую помощь в проведении съезда оказали руководство и члены оргкомитета, московские и уральские специалисты, ученые Екатеринбурга и, конечно, сотни молодых волонтеров (студентов и аспирантов) в красивых футболках с надписью “Я люблю химию”. С благодарностью надо отметить и гигантскую работу ученых секретарей съезда доктора химических наук, профессора Ю.Г. Горбуновой и кандидата химических наук О.А. Кузнецовой.
Местом проведения ХХ съезда был выбран город Екатеринбург – крупнейший центр химической и металлургической промышленности России, столица Урала.
Конечно же, читать, слушать рассказы, смотреть по телевидению весьма интересно, но, безусловно, ничто не заменит личное участие в съездах.
 Мне посчастливилось в жизни участвовать в нескольких Менделеевских съездах в Ленинграде, Минске, Москве, Волгограде… Самым незабываемым было участие в Х Менделеевском съезде в Ленинграде в 1969 г., который был посвящен 100-летию открытия Дмитрием Ивановичем Менделеевым периодической таблицы. Как говорится, с открытым ртом я смотрел на знаменитых организаторов и участников съезда, проводившегося в Таврическом дворце Ленинграда. В X съезде приняли участие 2000 человек, и в том числе много иностранных гостей. Назову несколько фамилий: Нобелевский лауреат Н.Н. Семенов, академики С.И. Вольфкович, Н.М. Жаворонков, В.И. Гольданский, И.В. Тананаев, Я.К. Сыркин, зам. председателя Совета министров В.А. Кириллин, министр образования В.П. Елютин и десятки известнейших ученых мира. А какие доклады сделали открыватели новых элементов – легенда науки академик Г.Н. Флеров (открыл 104 элемент – резерфордий, и в честь Флерова 114 элемент назван флеровий), председатель комиссии по атомной энергии США Г. Сиборг (синтезировал плутоний, а совместно с другими учеными открыл америций, кюрий, берклий, калифорний, эйнштейний, фермий, менделевий; в 1997 г. в честь Сиборга назван 106-й элемент – сиборгий). А доклады министра химической промышленности Л.А. Костандова, директора Института катализа академика Г.К. Борескова…
Мне посчастливилось в жизни участвовать в нескольких Менделеевских съездах в Ленинграде, Минске, Москве, Волгограде… Самым незабываемым было участие в Х Менделеевском съезде в Ленинграде в 1969 г., который был посвящен 100-летию открытия Дмитрием Ивановичем Менделеевым периодической таблицы. Как говорится, с открытым ртом я смотрел на знаменитых организаторов и участников съезда, проводившегося в Таврическом дворце Ленинграда. В X съезде приняли участие 2000 человек, и в том числе много иностранных гостей. Назову несколько фамилий: Нобелевский лауреат Н.Н. Семенов, академики С.И. Вольфкович, Н.М. Жаворонков, В.И. Гольданский, И.В. Тананаев, Я.К. Сыркин, зам. председателя Совета министров В.А. Кириллин, министр образования В.П. Елютин и десятки известнейших ученых мира. А какие доклады сделали открыватели новых элементов – легенда науки академик Г.Н. Флеров (открыл 104 элемент – резерфордий, и в честь Флерова 114 элемент назван флеровий), председатель комиссии по атомной энергии США Г. Сиборг (синтезировал плутоний, а совместно с другими учеными открыл америций, кюрий, берклий, калифорний, эйнштейний, фермий, менделевий; в 1997 г. в честь Сиборга назван 106-й элемент – сиборгий). А доклады министра химической промышленности Л.А. Костандова, директора Института катализа академика Г.К. Борескова…
Но вернемся в Екатеринбург. От имени моих соавторов – специалистов “НИАП-КАТАЛИЗАТОР”, НАК “Азот”, “Нижнекамскнефтехим”, я представил на съезде два доклада (по катализаторам метанирования и очистки пропана), а на круглом столе “Актуальные проблемы азотной промышленности” (под руководством чл.-корр. РАН В.Г. Систера) доклад о промышленных катализаторах, разработанных и производимых в “НИАП-КАТАЛИЗАТОР”.
Десятки секций и круглых столов, разбросанных в гигантском центре Екатеринбург-ЭКСПО, тысячи докладов прослушать, конечно, было невозможно. Назову лишь несколько самых впечатляющих.
Д.И. Менделеев много лет тому назад говорил: “Периодическая законность первая дала возможность видеть неоткрытые еще элементы в такой дали, до которой невооруженное этой законностью химическое зрение до тех пор не достигало, и при этом новые элементы ранее их открытия рисовались с целой массой свойств”. Эти предсказания вскоре блестяще оправдались. И в наши дни открытие новых элементов продолжается. Этому свидетельствует доклад Ю.Ц. Оганесяна и С.Н. Дмитриева об открытии новых сверхтяжелых элементов с атомными номерами 113 – 118 периодической таблицы Д.И. Менделеева. Открытие новой области стабильности сверхтяжелых элементов поставило целый ряд новых вопросов, и в частности, где границы периодической таблицы Д.И. Менделеева. Академик Юрий Оганесян вместе с двумя американскими учеными был номинирован на Нобелевскую премию по химии 2016 г., но премия досталась другим ученым. Наконец вопрос решен, и один из вновь открытых элементов теперь будет носить имя Оганесяна. Также решен вопрос о присвоении имени одному из новых элементов – московий (в честь Московского региона (г. Дубна), где расположен Объединенный институт ядерных исследований, в котором совместно с Ливерморской национальной лабораторией (США) были проведены эксперименты для обнаружения новых элементов). Правда, как заметил однажды академик Оганесян: “Открытия у нас делают одни, а право присвоить названия принадлежит другим”.
По наиболее близкой мне тематике (катализаторы) наибольшее впечатление произвели доклады академика РАН В.И. Бухтиярова, профессора В.М. Капустина, академика А.Г. Дедова, чл.-корр. З.Р. Исмагилова. Упоминаю только несколько докладов (из множества), которые лично удалось прослушать.
Значимость съезда была подчеркнута участием президента РАН академика В.Е. Фортова с докладом “Химические элементы в экстремальных условиях”. Речь в докладе шла о поведении химических элементов в условиях сверхвысоких давлений. При этом резко меняются их свойства. В ближайшие годы возможно и открытие химических элементов, не вписывающихся в таблицу Менделеева, что, несомненно, заставит расширить границы познания.
Безусловно, блестящий доклад сделал Нобелевский лауреат Даниэль Шехтман – профессор “ТЕХНИОН”
Хотел бы отметить и проблемы с аудиторией наших докладчиков. Требование времени – внедрение разработок, а производственников (техноруков, начальников цехов, ведущих и главных инженеров) крайне мало. Причина – недопонимание собственников и руководства предприятий, недостаточная информированность заводов и непомерная стоимость практически всех научных конференций, проводимых, по крайней мере, последние 15 – 20 лет. Наш съезд также не исключение, а ведь он посвящен как фундаментальным работам, так и прикладной химии. Отсюда во многом невостребованность российской науки, хотя есть и много других глобальных причин.
На заключительном заседании было отмечено, что в докладах и материалах съезда нашли отражение современные направления развития химической науки и техники, многие актуальные разработки, намечены перспективы их развития и использования. Работа съезда еще раз подтвердила, что химия занимает особое положение в процессе перехода нашей страны к устойчивому развитию, позволяя решать широкий круг задач – от изучения молекулярных основ жизни и факторов устойчивости окружающей природной среды до создания новых материалов и источников энергии.
Съезд продемонстрировал достижения российской и мировой химической науки в таких областях, как инновационные разработки по созданию новых материалов и технологий, включая наноматериалы и нанотехнологии, создание новых лекарств, электрохимическая энергетика, альтернативные энергоносители и моторные топлива из растительного сырья, экологически безопасная (т.н. зеленая) химия и проблемы устойчивого развития. Особое внимание было уделено вопросам химического образования и просвещения, а также борьбы с хемофобией.
Съезд обратил внимание Правительства страны и Комиссии по модернизации при президенте РФ на недопустимость механического слияния университетов, академических институтов.
Съезд считает, что одним из важнейших социальных факторов успешной реализации провозглашаемой руководством РФ стратегии модернизации экономики России на основе инноваций является наличие высококвалифицированных научных и инженерно-технических кадров. Подготовка таких кадров требует усиления внимания общества к среднему и высшему техническому образованию, а также к школьному образованию, призванному вызвать интерес учащихся к изучению естественнонаучных предметов – математики, химии, физики, биологии. В этой связи участники Менделеевского съезда считают необходимым просить Министерство образования и науки РФ обеспечить разработку и реализацию системы мероприятий по совершенствованию учебных программ (в том числе при увеличении количества часов на изучение химии и физики в школе) и методик преподавания химии и физики в средней школе.
Вместе с тем, несмотря на определенные успехи, съезд считает, что дальнейшее развитие химической науки и промышленности, химического образования и смежных отраслей, с учетом их значимости и потенциальных возможностей, требует принятия неотложных мер по ускорению коммерциализации результатов фундаментальных исследований и совершенствованию законодательной базы в вопросах интеллектуальной собственности. Необходимыми являются также обеспечение притока инвестиций, обновление технологий и инженерного обеспечения, расширение привлечения талантливой молодежи в химическую науку и промышленность, усиление ее социальной поддержки.

Кратко выскажу и свое мнение, которое излагал в ряде публикаций о работе отраслевых и академических институтов и вузов. Не способствовала успехам науки непродуманная реорганизация Академии наук, странное объединение вузов, сокращение сотрудников, низкое финансирование, превращение образования в школах и институтах в услуги. Напомню всем высказывание, приписываемое канцлеру Бисмарку: “Войну выигрывает школьный учитель”. По соотношению затрат на науку и ВВП Россия на последнем месте среди крупнейших развитых стран. Гибельным для российской науки является нижайший престиж ученых в последние годы. В Канаде проводился опрос: “Какое достижение больше поднимает патриотический дух – Нобелевская премия или число медалей на Олимпийских играх”? Почти 75 % канадцев выбрали Нобелевскую премию. У нас же, как писала газета “Известия”, олимпийский Сочи важнее Стокгольма.
Исчезновение с карты России из 6000 более 5000 отраслевых НИИ и КБ во многом сократило и возможности по внедрению разработок академических НИИ и вузов в промышленность.
В рамках съезда несколько часов было отведено VIII отчетно-выборному съезду Российского химического общества им. Д.И. Менделеева (РХО).
I съезд РХО состоялся в 1991 году в Ростове после распада СССР. Я был одним из делегатов съезда. Был принят Устав и президентом Российского химического общества стал академик Ю.А. Золотов. В последующие годы президентом Общества избирались академики А.И. Русанов, П.Д. Саркисов, А.Ю. Цивадзе.
В работе VIII съезда приняли участие делегаты из всех региональных организаций, и в том числе от Тульской организации химического общества – автор этих строк.
С докладом выступил президент РХО им. Д.И. Менделеева академик А.Ю. Цивадзе. Отметил сложное положение практически всех общественных организаций, отсутствие помощи руководства регионов, министерств, отсутствие помещений, плохое финансирование. Об этом же он говорил и при открытии ХХ Менделеевского съезда в присутствии двух тысяч участников, руководителей РАН и ФАНО.
После распада СССР РХО им. Д.И. Менделеева существует на весьма скромный бюджет из минимальных членских взносов. Причина – статус общественной организации, отсутствие финансирования от предприятий, вузов, НИИ… В зарубежных химических обществах ситуация иная. Например, в Великобритании химическое общество – это профессиональная структура, в которой состоит около 55000 членов. У общества большой бюджет, позволяющий выделять средства на премии, стипендии, издание журналов. Необходимо срочно оздоровлять деятельность Российского химического общества.
Были утверждены предложения по изменению Устава, касающиеся дифференцированного членства с соответствующими вступительными и ежегодными членскими взносами. Кроме того, теперь членами Общества могут быть, наряду с общественными организациями, и юридические лица. Президиуму Общества было поручено сформулировать предложения о Попечительском совете. Решено учредить золотую медаль РХО им. Д.И. Менделеева, которая будет присуждаться за выдающиеся заслуги в области химических наук и техники.
Конечно, не все благостно в деятельности РХО. Крайне мало молодых членов Общества и, на мой взгляд, большой потерей является приостановка издания популярного Бюллетеня “Химия в России”.
Президентом Общества вновь был избран директор института физической химии и электрохимии РАН, академик-секретарь отделения химии и наук о материалах РАН А.Ю. Цивадзе, а ученым секретарем к.б.н. Н.Р. Косинова. Были избраны вице-президенты, Президиум РХО и члены Правления.
Приятным и многообещающим заключением съезда явилось сообщение, что в 2019 году Менделеевский съезд будет посвящен 150-летию открытия периодического закона и 150-летию образования Русского химического общества (ранее Русское физико-химическое общество). Съезд пройдет в Санкт-Петербурге, где эти знаменательные события и произошли.
В Санкт-Петербурге располагается около 200 музеев, и все же позволю дать совет будущим участникам юбилейного съезда и, безусловно, всем, кто посетит Санкт-Петербург до знаменательного форума – посетить музей Дмитрия Ивановича Менделеева. Музей находится в историческом здании Двенадцати коллегий Санкт-Петербургского государственного университета. Музей был основан в 1911 г. по инициативе участников II Менделеевского съезда и находится в бывшей казенной квартире Дмитрия Ивановича, где он прожил почти четверть века, будучи профессором и зав. кафедрой университета.
П о приглашению Д.И. Менделеева студенты могли прийти на консультации, не выходя из здания университета, через дверь, ведущую в квартиру профессора. Сохранился кабинет ученого, собранная им огромная библиотека, многие из книг которой были вручены ему коллегами и друзьями. Наиважнейшую роль играет архив ученого. В этой квартире устраивались научные и художественные “среды”, на которых бывали: скульптор Петр Клодт, художники Иван Крамской, Архип Куинджи, Илья Репин, Иван Шишкин, музыкальный критик, историк искусств Владимир Стасов, композитор Александр Бородин, физиолог Иван Сеченов, адмирал Степан Макаров, поэт Александр Блок (зять Менделеева). Музей располагает прекрасной коллекцией картин, которые собирал Д.И. Менделеев. Сохранились: конторка, за которой Менделеев изобретал свой периодический закон, над которым работал 25 лет; его старинный фотоаппарат; приборы, на которых он проводил исследования; карта с отмеченными городами, где побывал Менделеев, и скатерть с автографами знаменитых людей того времени. Музей ежегодно посещают тысячи людей, среди которых были Нобелевские лауреаты И. Пригожин, Г. Сиборг, президент Франции Жак Ширак, знаменитые российские и зарубежные ученые.
о приглашению Д.И. Менделеева студенты могли прийти на консультации, не выходя из здания университета, через дверь, ведущую в квартиру профессора. Сохранился кабинет ученого, собранная им огромная библиотека, многие из книг которой были вручены ему коллегами и друзьями. Наиважнейшую роль играет архив ученого. В этой квартире устраивались научные и художественные “среды”, на которых бывали: скульптор Петр Клодт, художники Иван Крамской, Архип Куинджи, Илья Репин, Иван Шишкин, музыкальный критик, историк искусств Владимир Стасов, композитор Александр Бородин, физиолог Иван Сеченов, адмирал Степан Макаров, поэт Александр Блок (зять Менделеева). Музей располагает прекрасной коллекцией картин, которые собирал Д.И. Менделеев. Сохранились: конторка, за которой Менделеев изобретал свой периодический закон, над которым работал 25 лет; его старинный фотоаппарат; приборы, на которых он проводил исследования; карта с отмеченными городами, где побывал Менделеев, и скатерть с автографами знаменитых людей того времени. Музей ежегодно посещают тысячи людей, среди которых были Нобелевские лауреаты И. Пригожин, Г. Сиборг, президент Франции Жак Ширак, знаменитые российские и зарубежные ученые.
Съезд обратился к президенту РАН и в Министерство иностранных дел России, Международные организации с просьбой объявить 2019 год Международным годом Периодической таблицы химических элементов.
Пожелаем удачи будущим участникам мирового события.
Bis-thiourea compound helps stereospecifically build β-glycoside sugar linkages
Assembling oligosaccharides and polysaccharides can be tricky business. The steric and electronic elements that come into play when hooking one sugar onto another makes the synthesis of these molecules unpredictable. Now, inspired by the enzymes that connect sugars to one another in cells, Eric N. Jacobsen and coworkers at Harvard University have developed a macrocyclic catalyst that predictably and reliably builds certain types of sugar linkages—known as β-glycosidic bonds—in a stereospecific manner (Science 2017, DOI: 10.1126/science.aal1875).
Jacobsen’s group was hoping to use its expertise in chiral catalysts to influence the stereochemical outcome of a substitution reaction of a glycosyl chloride to create oligosaccharides. But the team discovered that when using a macrocyclic bis-thiourea catalyst, both enantiomers of the catalyst led to the same stereochemical outcome in the product.
Instead of the catalyst controlling the stereochemistry of the reaction, the stereochemistry of the sugar controls the reaction, Jacobsen explains. The reaction takes place via an SN2 mechanism: The thiourea groups in the catalyst make hydrogen bonds to the chloride on the sugar to make it a better leaving group, while an amide side chain in the catalyst activates an alcohol to make it a better nucleophile. The team used the catalyst to assemble a wide variety of trans-1,2-, cis-1,2-, and 2-deoxy-β-glycosides.
“There’s no reliable, general way to attach any two sugars together in a predictable way,” Jacobsen says. “That’s key if we’re going to help advance the field of oligosaccharide synthesis, including automated methods, for biological applications.” This new catalyst, he says, is a step in that direction.
“This is a fascinating study that links concepts from numerous parts of chemistry to define a new approach to selective carbohydrate synthesis,” says Scott J. Miller, an expert in organic synthesis at Yale University. “Achieving catalyst control with nonenzymatic catalysts, through the use of the reported bis-thioureas, opens up new doors and leverages the physical organic chemistry of the catalytic mechanisms in powerful new ways.”
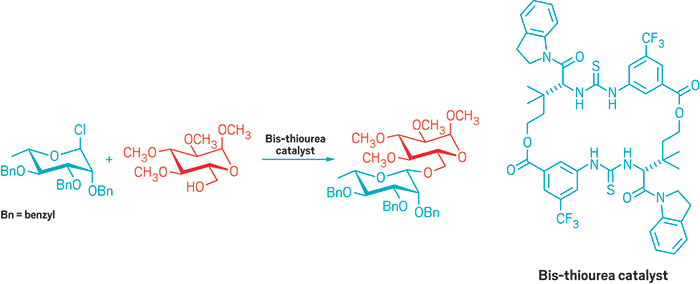
Carbohydrate connection: a macrocyclic bis-thiourea catalyst allows the Jacobsen group to create a ß-glycoside.
New method, dubbed MINFLUX, achieves nanometer resolution and fast analysis by combining the best of earlier imaging techniques
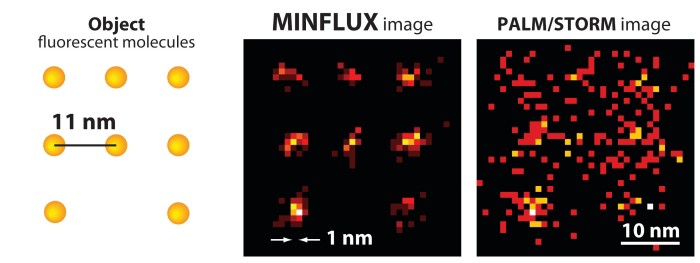
An array of fluorescent molecules (left) can be better resolved by MINFLUX (center) than by PALM/STORM (right).
Existing superresolution fluorescence microscopy methods such as STED and PALM have trouble imaging molecules with spatial resolution better than 20 to 40 nm. A new superresolution method called MINFLUX combines aspects of those earlier methods to resolve molecules just 6 nm apart with nanometer precision. The method is fast enough for short- and long-range tracking of individual molecules (Science 2016, DOI: 10.1126/science.aak9913).
STED and PALM (and the related technique STORM) distinguish densely packed features by allowing isolated molecules to fluoresce while those around them remain dark. The methods differ in how they determine the position of that fluorescence. In STED, a laser beam focused into a doughnut shape turns off fluorescence beneath the beam but not at the doughnut’s center. In PALM, fluorescent molecules are randomly switched on and off repeatedly so that only a few well-separated ones are activated at a time. The position of the activated molecules is determined with a camera.
“The strength of STED is that the doughnut always determines where the molecules are on and off and hence where the signal comes from,” says Stefan W. Hell of the Max Planck Institute for Biophysical Chemistry, who won the 2014 Nobel Prize in Chemistry for STED and leads the team that invented MINFLUX. On the other hand, “the strength of PALM is that you’reworking with individual molecules.” But the disadvantage of PALM is that those fluorescent molecules must emit many thousands of photons in order for scientists to get enough fluorescence intensity to determine their positions precisely.
In the new method, Hell, Francisco Balzarotti, also at the Max Planck Institute for Biophysical Chemistry, and coworkers usea doughnut-shaped laser beam for exciting fluorescence instead of turning it off. Because there’s no light intensity in the doughnut hole, a molecule located entirely within the hole won’t fluoresce. But a molecule that’s even slightly offset from the hole does fluoresce. How much it fluoresces depends on how far it is from the beam’s zero-intensity center. By scanning the doughnut in a defined pattern over the sample, the researchers can determine the precise position of a molecule from how its fluorescence emission changes.
“Because we can relate the position of the molecule to the ‘zero’ of the doughnut, many fewer photons are required to find out the position of the molecule than in PALM,” Hell says.
The team used a molecular assembly strategy called DNA origami to construct two arrays of fluorescent molecules at defined distances from one another: 11 nm in one array and 6 nm in the other. MINFLUX determined the molecules’ positions with a precision of 2.1 nm and 1.2 nm, respectively.
In addition to improved spatial resolution, the MINFLUX measurements are fast enough and require such low laser intensity that the researchers used the method for tracking individual molecules in live cells at multiple time scales.
“It’s a big deal that this method can be used for imaging but also for tracking at short and long time scales,” says Christy F. Landes, a superresolution expert at Rice University. “Not all methods are useful for these three very different imaging applications.”
Researchers create new reagents for a fluorinated version of copper-catalyzed azide-alkyne click chemistry
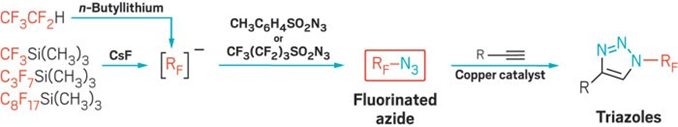
A two-step, one-pot reaction creates new fluorinated azides for copper-catalyzed fluorinated click chemistry.
With the help of new fluorinating reagents, a team led by Zsófia E. Blastik and Petr Beier of the Czech Academy of Sciences has devised a versatile fluorinated version of copper-catalyzed click chemistry. The approach overcomes some previous difficulties with preparing suitable fluorinated reagents for click reactions and opens the door to broader use of click chemistry, which has become invaluable to chemical biology and materials science.
The new chemistry hinges on the ability to make azidoperfluoroalkanes. Azidotrifluoromethane (CF3N3) has been known for some time, but its best reported synthesis starting from CF3I requires cumbersome handling of toxic and corrosive CF3NO, N2H4, and Cl2 gases—an approach that has limited CF3N3’s applications. Beier’s group alternatively attempted using electrophilic CF3I with sodium azide (NaN3) as a nucleophile, but the reaction didn’t work. Instead, the team found that CF3N3 and its previously unknown longer chain analogs can be prepared more conveniently from CF3Si(CH3)3 and related nucleophiles and sulfonyl azide electrophiles. In effect, the researchers flipped the polarity of the reaction’s constituents (Angew. Chem. Int. Ed. 2016, DOI: 10.1002/anie.201609715).
The reported azidoperfluoroalkanes undergo copper-catalyzed azide-alkyne cycloadditions, also known as click reactions, leading to N-perfluoroalkyl triazoles as underexplored building blocks, Beier says. “In fact, azidoperfluoroalkanes are more reactive in the click reaction with alkynes than nonfluorinated alkyl azides.”
“A reminder emerging out of this paper by Beier and colleagues is that, if you want to build a bond by a polar mechanism, there are always two options,” says Andrei K. Yudin of the University of Toronto, who focuses on the chemistry of heterocyclic compounds. “It is a good idea to reverse the polarity of components if one path is problematic. As a result, they have developed an efficient entry into azidoperfluoroalkanes.”
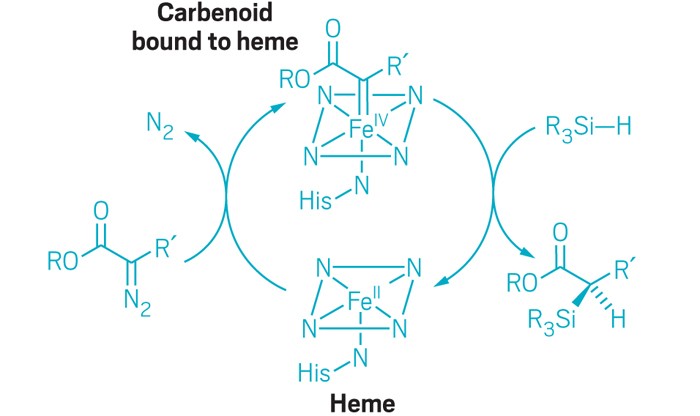
Bacterial cytochrome c demonstrates first example of biologically catalyzed organosilicon chemistry
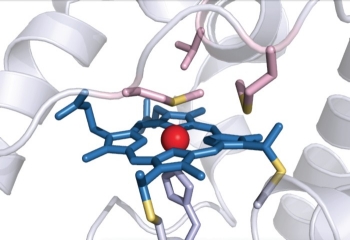 |
The heme group of R. marinus cytochrome c catalyzes carbenoid insertion into Si-H bonds. |
Silicon is the second most abundant element in Earth’s crust after oxygen, but carbon-silicon bonds are unheard of in nature: Neither biological organosilicon compounds nor biosynthetic pathways to create them have been identified. But when given the right starting materials, some heme proteins can stereospecifically form carbon-silicon bonds, report researchers from Caltech (Science 2016, DOI: 10.1126/science.aah6219).
“Nature’s iron heme chemistry just jumps on this opportunity because we provided it with the right precursors,” says Frances H. Arnold, who led the work with S. B. Jennifer Kan. “It’s a profound demonstration of how easily nature can innovate.”
 |
Three mutations of cytochrome c turn the protein into an enzyme that produces organosilicon compounds with greater than 99% enantiomeric excess. |
“This closes a crucial gap between biological and chemical catalysis,” writes Martin Oestreich of the Technical University of Berlin in a commentary accompanying the paper. “The impact is unforeseeable, but it seems that we are a big step closer to potentially facilitating industrially relevant reactions such as alkene hydrosilylation with biomolecules.”
Chemists have experimented for decades with forming chiral organosilicon compounds by carbenoid insertion into Si–H bonds, but the reactions typically require halogenated solvents and catalysts made of precious metals coordinated with chiral ligands. The catalysts also have low turnover—each catalyst complex survives fewer than 100 reactions before it inactivates.
Prior work in Arnold’s lab and elsewhere had demonstrated that heme proteins can catalyze nonnatural carbene transfer reactions through insertion into N–H and S–H bonds. In the new experiments, the researchers screened a panel of heme proteins to find ones that could catalyze insertion of ethyl 2-diazopropanoate into the Si–H bond of dimethyl(phenyl)silane. Cytochrome c from the bacterium Rhodothermus marinus, which is found in submarine hot springs in Iceland, catalyzed the reaction with 97% enantiomeric excess, although the catalytic turnover was still low.
Cytochrome c proteins normally don’t catalyze chemical reactions; instead they transfer electrons between biomolecules in cells. But that did not stop Kan, Arnold, and colleagues from pushing the R. marinus cytochrome c to improve its newfound ability to perform organosilicon catalysis.
The researchers used directed evolution to come up with a set of three mutations that together increase the new enzyme’s enantioselectivity to greater than 99% and turnover to greater than 1,500. One of the mutations involved a methionine residue that provides an axial ligand to the protein’s iron center. A change from methionine to aspartic acid possibly provided more substrate access for the formation of an iron-carbenoid intermediate.
The “evolved” cytochrome c performs stereoselective carbene insertion into Si–H bonds using a variety of silicon and diazo reagents, without competing cyclopropanation. And when given 4-(dimethylsilyl)aniline, which can accommodate insertion at both N–H and Si–H bonds, the enzyme formed the organosilicon product with 97% chemoselectivity. Arnold thinks that the partially exposed active site promotes substrate promiscuity but that there’s also some characteristic of the heme pocket that prefers silicon chemistry.
Chemical & Engineering News
ChemCatChem
Origin of the Improved Performance in Lanthanum-doped Silica-supported NiCatalysts
D. Baudouin, T. Margossian, U. Rodemerck, P.B. Webb, L. Veyre, F. Krumeich, J.-P. Candy, C. Thieuleux, C. Copére
DOI: 10.1002/cctc.201600582
Advanced Synthesis & Catalysis
Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions
H. Pellissier
DOI: 10.1002/adsc.201600462
Angewandte Chemie International Edition
Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and
Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry
J. Klankermayer, S. Wesselbaum, K. Beydoun, W. Leitner
DOI: 10.1002/anie.201507458
Chemistry – A European Journal
A One-Pot Synthesis of N-Aryl-2-Oxazolidinones and Cyclic
Urethanes by the Lewis Base Catalyzed Fixation of Carbon Dioxide into Anilines and Bromoalkanes
T.Niemi, J.E. Perea-Buceta, I. Fernández, O.-M. Hiltunen, V. Salo,S. Rautiainen, M.T. Räisänen, T. Repo
DOI: 10.1002/chem.201602338
ChemBioChem
Widening
the Product Profile of Carbon Dioxide Reduction by Vanadium Nitrogenase
J. G. Rebelein, Y. Hu, M.W. Ribbe
DOI: 10.1002/cbic.201500305
Chemistry – An Asian Journal
Silver(I)-Catalyzed
Three-Component Reaction of Propargylic Alcohols, Carbon Dioxide and Monohydric Alcohols: Thermodynamically Feasible Access to
β-Oxopropyl Carbonates
Zhi-Hua Zhou, Qing-Wen Song, Liang-Nian He et al
DOI: 10.1002/asia.201600600
ChemPlusChem
Electrocatalytic
Reduction of Carbon Dioxide with a Well-Defined PN3−Ru Pincer Complex
S. Min, S. Rasul, H. Li, D.C. Grills, K. Takanabe,L.-J. Li,K.-W. Huang
DOI: 10.1002/cplu.201500474
Advanced Materials Interfaces
Constructing
Ru/TiO2 Heteronanostructures Toward Enhanced Photocatalytic Water Splitting via a
RuO2/TiO2 Heterojunction and Ru/TiO2 Schottky
Junction
Q.Gu, Z. Gao, S. Yu, C. Xue
DOI: 10.1002/admi.201500631
Advanced Science
Visible and Near-Infrared Photothermal Catalyzed Hydrogenation of Gaseous
CO2 over Nanostructured Pd@Nb2O5
J.Jia, P.G. O'Brien, L. He, Q. Qiao, Teng Fei, L.M. Reyes, T.E. Burrow, Y. Dong, K. Liao, M. Varela, S.J. Pennycook, M. Hmadeh, A.S. Helmy, N.P. Kherani, D.D. Perovic, G.A. Ozin
DOI: 10.1002/advs.201600189
Energy
Technology
Enhancement
of the Efficiency and Selectivity for Carbon Dioxide
Electroreduction to Fuels on Tailored Copper Catalyst Architectures
M. Bevilacqua, J. Filippi, M. Folliero, A. Lavacchi,H.A. Miller, A. Marchionni, F. Vizza
DOI: 10.1002/ente.201600044
Macromolecular Chemistry and Physics
The
Synthesis of Poly(ethylene glycol) (PEG) Containing Polymers via
Step-Growth Click Coupling Reaction for CO2 Separation
B. N. Gacal, V.Filiz, V. Abetz
DOI: 10.1002/macp.201500381
Particle Systems Characterization
Progress
of Carbon Quantum Dots in Photocatalysis Applications
Z.Zhang, T. Zheng, X. Li, J. Xu, H. Zeng
DOI: 10.1002/ppsc.201500243
 |
Academician G.K. Boreskov |
|
Школа-конференция молодых ученых с международным участием “V Научные чтения, посвященные памяти академика А.Е. Фаворского” Иркутск, Россия |
http://www.irkinstchem.ru/index.php/ru/ ob-institute/conference |
|
6th International Conference on Ecological & Environmental Chemistry 2017 Chisinau, Republic of Moldova |
www.EEC-2017.mrda.md |
|
18th Netherlands' Catalysis and Chemistry Conference Noordwijkerhout, The Netherlands |
http://www.n3c.nl |
|
5th International Conference on Multifunctional, Hybrid and Nanomaterials Lisbon, Portugal |
http://www.hybridmaterialsconference.com |
|
Recent Developments in Synthesis and Catalysis (RDSC-2017) Dibrugarh, India |
http://www.dibru.ac.in/seminar/rdsc2017 |
|
50th Annual Meeting of German Catalysis (GECATS 50) Weimar, Germany |
http://dechema.de/en/katalytiker2017.html |
|
World Congress and Expo on Nanotechnology and Nanoengineering Dubai, United Arab Emirates |
https://biocoreconferences.com/nanotechnology2017 |
|
7th International Conference on Graphene and Two-dimensional Materials (GRAPHENE 2017) Barcelona, Spain |
http://www.grapheneconf.com/2017 |
|
2nd International Conference on Nanotechnology and Environmental Issues (ICNEI'17) Barcelona, Spain |
http://icnei.com |
|
3rd Energy and Materials Research Conference (EMR 2017) Lisbon, Portugal |
http://emr2017.org |
|
VIII научная конференция молодых ученых “Инновации в химии: достижения и перспективы” МГУ, Москва, Россия |
http://www.chem.msu.ru/rus/events/LomonosovCh2017 |
|
IV Scientific Conference “Boreskov Readings” dedicated to the 110th anniversary of Academician Georgii K. Boreskov Novosibirsk, Russia |
http://conf.nsc.ru/boreskov-IV/en |
|
VII Всероссийская конференция с международным участием “Новые достижения в химии и химической технологии растительного сырья” Барнаул, Россия |
http://konf.asu.ru/cprm-2017 |
|
XIV Международная конференция студентов, аспирантов и молодых ученых “Перспективы развития фундаментальных наук” Томск, Россия |
http://science-persp.tpu.ru |
|
Программа повышения квалификации “Современные тенденции в получении и исследовании функциональных материалов” Томск, Россия |
http://science-persp.tpu.ru +7-923-448-20-00 zykovaap@mail.ru |
|
2nd Green and Sustainable Chemistry Conference (2GSCC 2017) Berlin, Germany |
www.greensuschemconf.com/ |
|
III Всероссийская молодежная конференция “Достижения молодых ученых: химические науки” Уфа, Республика Башкортостан, Россия |
http://www.bashedu |
|
3-я Всероссийская молодежная научная конференция с международным участием “Экологобезопасные и ресурсосберегающие технологии и материалы” Улан-Удэ, Республика Бурятия, Россия |
ertm3@mail.ru тел. (3012) 433171 |
|
29th European Symposium on Applied Thermodynamics (ESAT 2017) Bucharest, Romania |
http://www.esat2017.ro |
|
IV Школа-конференция молодых ученых “Неорганические соединения и функциональные материалы” Новосибирск, Россия |
icfm.2017@gmail.com http://www.niic.nsc.ru/institute/conferences-inx/701-conferences-2017 |
|
III Российский конгресс по катализу “Роскатализ-2017” Нижний Новгород, Россия |
http://conf.nsc.ru/ruscatalysis-2017 |
|
25th North American Meeting (NAM) of the Catalysis Society Denver, Colorado, USA |
http://www.nam25.org |
|
Всероссийская научная конференция с международным участием “Современные проблемы органической химии” Новосибирск, Россия |
http://web.nioch.nsc.ru/conf2017 |
|
IX Международная конференция “Полимерные материалы пониженной горючести” Алматы, Республика Казахстан |
http://www.kaznu.kz/ru |
|
8th International IUPAC Symposium “Macro- and Supramolecular Architectures and Materials: Multifunctional Materials and Structures” (MAM-17) Sochi, Russia |
http://www.mam-17.org |
|
European Biomass Conference & Exhibition (EUBCE 2017) Stockholm, Sweden |
http://www.eubce.com |
|
VII Symposium on Hydrogen, Fuel Cells and Advanced Batteries (HYCELTEC 2017) Porto, Portugal |
hyceltec2017@fe.up.pt |
|
9-th International Symposium “Molecular Mobility and Order in Polymer Systems” Saint Petersburg, Russia |
http://www.mmops2017.com |
|
19th IUPAC International Symposium on Organometallic Chemistry Directed Towards Organic Synthesis (OMCOS 2017) Jeju Island, Korea |
www.omcos.org |
|
7th International Congress on Ionic Liquids (COIL 7) Mont Tremblant, Quebec, Canada |
http://www.engconf.org/conferences/chemical
-engineering/conference-on-ionic-liquids-coil |
|
Meeting of the Spanish Catalysis Society (SECAT'17) Oviedo, Spain |
http://www.secat17.com/en |
|
XXI International Conference on Chemical Thermodynamics in Russia (RCCT-2017) Novosibirsk, Russia |
http://rcct2017.ru |
|
Heterogeneous Catalytic Hydrogenation (a training course) Lisbon, Portugal |
http://www.rsc.org/events/detail/23259/ heterogeneous-catalytic-hydrogenation |
|
II Всероссийская научная конференция с международным участием “Актуальные проблемы адсорбции и катализа” Плес, Россия |
http://isuct.ru/conference/apaik |
|
VI Всероссийская школа- конференция молодых ученых “Органические и гибридные наноматериалы” Иваново, Россия |
http://ivanovo.ac.ru/science/research/item/1811 |
|
20th European Symposium on Organic Chemistry (ESOC 2017) Cologne, Germany |
http://www.esoc.uni-koeln.de |
|
22nd International Symposium on Olefin Metathesis and Related Chemistry (ISOM 2017) Zürich, Switzerland |
www.coperetgroup.ethz.ch/isom-22.html/ |
|
22nd EuCheMs Conference on Organometallic Chemistry (EuCheMs OMC 2017) Amsterdam, the Netherlands |
http://www.eucomc2017.amsterdam |
|
46th IUPAC World Chemistry Congress (IUPAC-2017) Sao Paulo, Brazil |
www.iupac2017.org/ |
|
9th International Seminar on Flame Structure (9ISFS) Novosibirsk, Russia |
http://www.kinetics.nsc.ru/kcp/9ISFS/index.htm |
|
Liverpool Catalysis Summer School Liverpool, Great Britain |
http://www.liv.ac.uk/chemistry/events/ catalysis-summer-school-2017 |
|
15th International Conference on Carbon Dioxide Utilization (ICCDU XV) Shnaghai, China |
http://iccdu2017.csp.escience.cn/dct/page/1 |
|
18th International Symposium on Relations between Homogeneous and Heterogeneous Catalysis (ISHHC XVIII 2017) Sydney, Australia |
http://catalysis.org.au/events/ishhc-18 |
|
8th International Conference on Green and Sustainable Chemistry (GSC-8) Melbourne, Australia |
www.racicongress.com/GSC8/ |
|
13th European Congress on Catalysis (Europacat 2017) Florence, Italy |
www.europacat2017.eu/ |
|
11th Triennial Congress of the World Association of Theoretical and Computational Chemists (WATOC 2017) Munich, Germany |
http://www.watoc2017.com |
|
XIX Euroanalysis Conference Stockholm, Sweden |
http://euroanalysis2017.se |
|
15th International Conference on Environmental Science and Technology (CEST 2017) Rhodes, Greece |
http://cest.gnest.org |
|
8th World Congress on Oxidation Catalysis “Oxidation processes: challenges and solutions” (WCOC 2017) Krakow, Poland |
http://8wcoc.icsc.pl |
|
XII European Workshop Meeting on Innovation in Selective Oxidation (ISO’17) Krakow, Poland |
http://8wcoc.icsc.pl |
|
IV International Conference “Catalysis for Renewable Sources: Fuel, Energy, Chemicals” (CRS-4) Gabicce Mare, Italy |
http://conf.nsc.ru/CRS4/en |
|
XXIX Симпозиум “Современная химическая физика” Туапсе, Россия |
www.chemphysics.ru |
|
Международная конференция “Возобновляемые растительные ресурсы: химия, технология, медицина” (Renewable Resources RR 2017) Санкт-Петербург, Россия |
http://onlinereg.ru/RR2017 |
|
7th IUPAC Conference on Green Chemistry (7th ICGC) Moscow, Russia |
http://greeniupac2017.muctr.ru |
|
IX Научно-практическая конференция с международным участием “Сверхкритические флюиды: фундаментальные основы, технологии, инновации” Сочи, Россия |
http://conf.scftec.ru |

