
Поздравляем лауреатов
Терминология в гетерогенном катализепод ред. Р.П.Бурвелла
(окончание )
Русский перевод
Ориганальная версия
Конференция памяти Ю.И.Ермакова
Международное рабочее совещание "Происхождение и эволюции биосферы"
2-я Международная Школа-конференция молодых ученых по катализу
7-й Европейский конгресс по катализу
За рубежом
Приглашения на конференции
|
Поздравляем лауреатов премии имени В.А. Коптюга - Президиум Российской академии наук присудил премию имени В.А. Коптюга 2005 года к.т.н. Андрею Николаевичу Загоруйко, д.х.н. Зинферу Ришатовичу Исмагилову и академику Геннадию Викторовичу Саковичу за работу "Разработка и внедрение новых каталитических технологий охраны окружающей среды и утилизации высокоэнергетических материалов". Авторы разработали ряд высокоэффективных катализаторов и технологий обезвреживания жидких и газообразных отходов. Совместно с предприятиями Росатома предложена технология компактирования органических радиоактивных отходов, основанная на каталитическом окислении. Достигнуты успехи в технологии окисления паров гептила, обеспечившие соблюдение санитарных норм при проведении регламентных работ с топливом. Решена задача защиты персонала и окружающей среды. Для твердых ракетных топлив создана технология, включающая химическое разрушение полимерного связующего и дальнейшее выделение ценных компонентов. | ||
 |
 |
 |
1.6 Транспортные явления в гетерогенном катализе
В данном разделе не будет сделана попытка описать больше технических аспектов технологии химических реакторов.
Отличительной чертой гетерогенных химических реакций является легкость, с которой законы химической кинетики маскируются различными транспортными явлениями, связанными с существованием градиентов концентрации и/или температуры в пограничном гидродинамическом слое, окружающем частицы катализатора (внешние градиенты) или в пористой текстуре самих частиц катализатора (внутренние градиенты). Дополнительные трудности возникают в реакторах периодического действия и в проточных реакторах с перемешиванием, если смешивание недостаточно для поддержания однородных концентраций в жидкой фазе. Перемешивание особенно важно там, где один из реагентов - газ, а катализатор и другие реагенты и продукты находятся в конденсированных фазах, например, при гидрировании жидкого алкена. Здесь перемешивание должно обеспечивать фугитивность растворенных газообразных реагентов, равную их фугитивности в газовой фазе.
Если внешние градиенты таковы, что существует значительная разница между концентрацией или температурой в объеме жидкости и концентрацей или температурой на внешней поверхности катализатора, скорость реакции на поверхности значительно отличается от той, которая преобладала бы, если бы они были равны. Тогда говорят, что на каталитическую реакцию влияет внешний массо- или теплоперенос, соответственно, а когда это влияние преобладает, то скорость соответствует режиму внешнего массо - или теплопереноса.
Аналогично, если внутренние градиенты концентрации или температуры различаются на внешней поверхности частицы катализатора и в ее центре, скорость в частице существенно отличается от скорости, которая преобладала бы, если бы концентрация или температура были одинаковы по всей частице. Тогда говорят, что на каталитическую реакцию влияет внутренний массо- или теплоперенос, а когда это влияние преобладает, то скорость соответствует режиму внутреннего массо - или теплопереноса.
Такие термины, как лимитируемая диффузией или контролируемая диффузией нежелательны, потому что скорость в режимах тепло- или массопереноса может быть больше, чем в кинетическом режиме работы, т.е. когда градиенты пренебрежимо малы.
1.7 Потеря каталитической активности
1.7.1 Отравление и ингибирование
Следы примесей в жидкости, с которой соприкасается катализатор, могут адсорбироваться на активных центрах и уменьшать или прекращать каталитическое действие. Это явление называют отравлением, а воздействующую таким образом примесь - ядом. В случае сильной и практически необратимой адсорбции яда отравление называется постоянным. Если адсорбция яда слабее и обратима, удаление яда из жидкой фазы приводит к восстановлению первоначальной каталитической активности. Такое отравление называется временным. Если адсорция яда еще слабее и не сильно преобладает над адсорбцией реагента, уменьшение скорости под действием яда можно назвать конкурентным ингибированием или ингибированием. Конечно, яда может быть гораздо больше, чем следовые количества. И конечно же, нет четких границ в последовательности: постоянное отравление, временное отравление, конкурентное ингибирование.
При селективном отравлении или селективном ингибировании яд тормозит скорость одной катализируемой реакции больше, чем скорость другой или может затормозить только одну реакцию. Например, существуют яды, которые тормозят гидрирование олефинов гораздо сильнее, чем гидрирование ацетиленов или диенов. И оказалось, что следы соединений серы селективно ингибируют гидрогенолиз углеводородов во время каталитического риформинга.
Продукт реакции также может быть ингибитором или ядом. Это явление называется самоотравление или автоотравление.
1.7.2 Дезактивация: общие положения
Конверсия в каталитической реакции, проводимой при постоянных условиях реакции, часто уменьшается со временем пробега или временем действия. Это явление называется дезактивацией катализатора или распадом катализатора. Если есть возможность определить кинетическую форму реакции и, таким образом, измерить константу скорости k каталитической реакции, то иногда становится возможным выразить скорость дезактивации эмпирической формулой.
где t - время действия, n - некая положительная константа, а B остается константой в процессе реакции, но зависит от температуры и других условий реакции. Кроме того, можно предположить, что k уменьшается в результате исчезновения активных центров, и k может быть заменено на L в предыдущем уравнении, где L рассматривается как эффективная концентрация поверхностных центров. Кроме того, обычно определяется время дезактивации (или время распада) как время реакции, в течение которого k падает до определенной доли ее первоначальной величины, часто 0,5. Времена дезактивации могут меняться от минут, как в каталитическом крекинге, до лет, как в гидрообессеривании.
Дезактивация катализатора иногда может быть обратимой, и исходная активность катализатора восстанавливается с помощью определенных операций, называемых регенерацией. Например, закоксованный катализатор крекинга регенерируется с помощью выжигания кокса (см. §§1.7.3., 1.9).
Если каталитическая реакция включает совокупность различных процессов, дезактивация может привести к изменению распределения продуктов. В таких случаях дезактивация не только снижает суммарную скорость, но и изменяет селективность.
1.7.3 Типы дезактивации
Причиной дезактивации катализатора может быть дезактивация каталитических центров из-за отравления примесями или продуктами каталитической реакции (§1.7.1). Многие реакции с участием углеводородов, особенно протекающие при повышенных температурах, приводят к отложению на катализаторе высокомолекулярных соединений углерода и водорода, которые дезактивируют катализатор. Это явление называется закоксовывание или загрязнение. Такие дезактивированные катализаторы зачастую могут быть регенерированы.
Дезактивация может быть и результатом изменения структуры или текстуры катализатора. Изменения такого рода обычно необратимы, и катализатор не может быть регенерирован. Этот тип дезактивации часто называют старением катализатора.
Спекание и перекристаллизация. Катализаторы во время использования часто страдают от постепенного увеличения среднего размера кристаллитов или роста первичных частиц. Это обычно называют спеканием. Спекание приводит к уменьшению площади поверхности и, следовательно, к уменьшению числа каталитических центров. В некоторых случаях спекание приводит к изменению каталитических свойств центров, например, для катализатора, состоящего из высокодисперсного металла на носителе каталитические свойства могут изменяться при спекании за счет изменения сравнительной доступности различных кристаллических граней металлического компонента катализатора или по какой-либо иной причине. Таким образом, спекание приводит к снижению скорости, а возможно, и к изменению селективности. Подобные явления могут наблюдаться и для окисного катализатора, применяемого для каталитического окисления. Размер кристаллов увеличивается, или изменяется первоначальная структура кристаллов. Например, твердые бинарные соединения могут разлагаться на компоненты, или аморфная масса может кристаллизоваться. Эти процессы можно назвать перекристаллизацией. В некоторых случаях "спекание" и "перекристаллизация" используются для обозначения одних и тех же процессов. Эти процессы могут сопровождаться исчезновением поверхностных дефектов.
В некоторых случаях, как, например, в каталитическом крекинге на алюмосиликатах, процессы, подобные тем, что происходят при спекании и перекристаллизации, могут привести к изменению текстуры катализатора. Уменьшается поверхность и изменяется распределение пор по размерам.
1.8 Механизм каталитических реакций
1.8.1 Общие положения
Химическая реакция включает ряд элементарных процессов (Руководство, §11.3), которые могут протекать последовательно или параллельно. Эти процессы начинаются и заканчиваются при минимуме свободной энергии компонентов (реагентов, интермедиатов и продуктов), а каждый элементарный процесс проходит через состояние с максимумом свободной энергии (переходное состояние). Для определения механизма необходимо определить элементарные процессы. Таким образом определяются интермедиаты. Нужно также знать природу (энергетику, структуру, распределение зарядов) переходного состояния. Это все так же, как в химии вообще. Особенность механизма в гетерогенном катализе - это то, что он включает реакции между сорбированными веществами и активными центрами, реакции между адсорбатами и процессы регенерации активных центров по типу цепной реакции.
В общем случае возможна только частичная реализация вышеописанного подхода к определению механизма реакции.
Иногда термин "механизм" используется в разных смыслах. Например, рассмотрим два случая.
|
A + B = C |
vs |
A + B |
|
C |
C = D |
Можно сказать, что в этих двух случаях реализуются разные механизмы или что это два варианта одного и того же механизма.
1.8.2 Элементарные процессы в гетерогенном катализе
В гетерогенном катализе существует намного больше типов элементарных процессов, чем в газофазных реакциях. Элементарные процессы гетерогенного катализа в общем случае разделяются на процессы адсорбции-десорбции или поверхностные реакции, т.е. элементарные процессы, включающие реакции адсорбированных групп. Свободные поверхностные центры и молекулы могут участвовать, а могут и не участвовать в стадиях поверхностной реакции.
Не существует общепринятой классификации элементарных процессов гетерогенного катализа. Однако общеприняты названия нескольких типов элементарных процессов, и получила некоторое хождение частная классификация [см. M. Boudart, Kinetics of Chemical Processes, Chap. 2 (1968)]. Реакции, приведенные ниже в качестве примеров такой терминологии, были предложены в литературе, но не установлено точно, все ли они происходят в природе с какой-либо заметной скоростью.
Адсорбция-десорбция включает процессы физической адсорбции, а также недиссоциативной хемисорбции.
* + NH3(г) 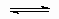 H3N*
H3N*
* + H(г) H*
Диссоциативная адсорбция и ее противоположность, ассоциативная десорбция.
2* + CH4(г) CH3* + H*
Можно предположить, что метан реагирует или из газовой фазы, или из физосорбированного состояния.
Диссоциативная поверхностная реакция и ее противоположность, ассоциативная поверхностная реакция.
2* + C2H5* H* + *CH2CH2*
Она включает "диссоциативную адсорбцию" адсорбата.
Сорбционное внедрение - это аналог процесса внедрения лиганда в координационной химии.
H* + C2H4(g) 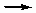 *C2H5
*C2H5
Эту реакцию также можно представить как реакцию, протекающую с адсорбцией C2H4 и последующим переходом лиганда (поверхностная ассоциативная реакция).
Реактивная адсорбция и ее противоположность, реактивная десорбция, похожа на диссоциативную адсорбцию, но один фрагмент присоединяется к адсорбату, а не к поверхностному центру.
В процессах отщепления и экстракции группа адсорбтива или адсорбата забирает, соответственно, адсорбированный атом или атом из решетки.
Процесс отщепления *H + H(g) * + H2(g)
Следующий элементарный процесс, происходящий на одном или, как показано, на двух центрах, называется механизмом Риделя или Риделя-Элея:
|
D-D(g) |
H - D(g) | |
|
H |
D |
|
|
* * |
|
* * |
D2 можно рассматривать и как некую разновидность слабо адсорбированного состояния. Заметим, что один атом D никогда не образует с поверхностью интермедиат с минимальной энергией Гиббса. Именно для таких процессов рекомендован термин "механизм Риделя или Риделя-Элея". Однако он использовался и для аналогичных процессов, в которых молекула реагента и молекула продукта с примерно одинаковой энергией находились в жидкой фазе или в некотором слабо адсорбированном состоянии и в которых один или больше атомов никогда не образовывают связи с поверхностью. Примером может быть следующий элементарный процесс ,который назвали свитч-процессом. Такой термин мог бы использоваться как общее название подобных процессов. Позднее термин "механизм Риделя или Риделя-Элея" был распространен на все элементарные процессы, в которых молекула реагирует из жидкой фазы или из слабо адсорбированного состояния. Даже вышеупомянутые процессы сорбционного внедрения и отщепления попадают в круг такого расширенного определения.
1.8.3 Номенклатура поверхностных интермедиатов
Названия поверхностных интермедиатов, пока это возможно, соответствуют общей химической номенклатуре.
Адсорбированные группы могут рассматриваться как поверхностные соединения, аналогичные молекулярным соединениям. Например, *H можно назвать поверхностным гидридом, *=C=O - линейным поверхностным карбонилом, а мостиковым поверхностным карбонилом. H2N* можно назвать поверхностным амидом, а H3C* - поверхностным метилом или поверхностным s -алкилом.
Группа *H также может быть названа адсорбированным водородом, а *CO - адсорбированным оксидом углерода.
Особую проблему представляют органические адсорбаты, поскольку постоянно обсуждаются довольно сложные специфические структуры. Рекомендована номенклатура, в которой поверхность рассматривается как заместитель, который замещает один или более атомов водорода. Степень замещения указывается как моноадсорбированный, диадсорбированный и т.д. Такая терминология не определяет ни природу химической связи с поверхностью, ни ограничивает a priori валентность поверхностного центра *. Таким образом, обе группы называются 1,3-диадсорбированный пропан. Другие примеры приведены ниже:
|
*CH3 |
моноадсорбированный метан |
|
*CH2CH2CH3 |
1-моноадсорбированный пропан |
|
*OCH2CH3 |
O-моноадсорбированный этанол |
|
*CH2CH2OH |
2-моноадсорбированный этанол |
|
* =CH-CH3 или (*)2CH-CH3 |
1,1-диадсорбированный этан |
|
* =NH или (*)2NH |
диадсорбированный аммиак |
|
*COCH3 |
1-моноадсорбированный |
Номенклатурную систему замещения следует рассматривать как такую, которая показывает только, как атомы связаны с поверхностью, но не определяет точную электронную структуру. Таким образом, p -адсорбированный этилен - это представленный другим образом 1,2-диадсорбированный этан.
Не стоит придерживаться номенклатуры, базирующейся на процессе образования конкретного адсорбата. Таким образом, *H может быть "диссоциативно адсорбированным водородом", но такие же группы образуются при диссоциативной адсорбции CH4, NH3, H2O.
1.9 Номенклатура каталитических реакций
В общем случае название каталитической реакции может образовываться добавлением прилагательного "каталитическая" к обычному химическому названию реакции, например каталитическое гидрирование (или, если требуется внести ясность, гетерогенное каталитическое гидрирование каталитическое гидрообессеривание, каталитическое окислительное дегидрирование ,каталитическая стереоспецифическая полимеризация.
В общем случае не используется специальная терминология для реакций. Однако определенные каталитические процессы, представляющие интерес с точки зрения технологии, имеют свои собственные общеупотребимые названия. Там, где такие процессы предполагают одновременное протекание двух или более химических реакций, применение таких специальных названий неизбежно. Ниже даны примеры таких важных технологических процессов:
Каталитический крекинг. В этом процессе высококипящая фракция нефти, например, нефтяной газ, превращается, в основном, в более низкокипящий материал с высоким октановым числом. Из реакций, которые оказываются вовлеченными в процесс, можно назвать скелетную изомеризацию алканов с их последующим разложением на алкан и олефин и реакции переноса водорода, уменьшающие количество образующегося олефина и приводящие к образованию кокса и ароматических углеводородов.
Каталитический гидрокрекинг. Эта реакция похожа на промышленный каталитический крекинг, но проводится под давлением водорода и на катализаторе, содержащем компонент с гидрирующим действием.
Каталитический риформинг. Каталитический риформинг - это процесс, проводимый с целью увеличения октанового числа нафты. Он включает изомеризацию алканов, дегидрирование циклогексанов в ароматические углеводороды, изомеризацию или дегидрирование алкилциклопентанов и дегидроциклизацию алканов.
Можно упомянуть следующие реакции, являющиеся редким исключением среди гетерогенных каталитических реакций и имеющие особое значение в катализе.
Каталитическое метанирование. Это процесс удаления моноокиси углерода из потока газов или получения метана по реакции
CO + 3H2 CH4 + H2O
Каталитическая дегидроциклизация. Это реакция, в которой алкан превращается в ароматический углеводород и водород, например,
гептан толуол + 4H2
Каталитический гидрогенолиз. Это обычно используемая реакция, в которой º C-Cº + H2 дает º CH + HCº , например,
пропан + H2 этан + метан
толуол + H2 бензол + метан
бутан + H2 2 этан
Однако он также может использоваться и для разрыва связей, отличных от ºC-Cº, например,
бензилацетат + H2 толуол + уксусная кислота
бензиламин + H2 толуол + NH3
Каталитическое гидрообессеривание. Это процесс, в котором в присутствии водорода сера удаляется в виде сероводорода.
| 2.1 Catalysis and catalysts | 2.1 Катализ и катализаторы | |
| 2.2 Adsorption | 2.2 Адсорбция | |
| Area of surface | A, As, S | Площадь поверхности |
| Specific surface area | a, as, s | Удельная площадь поверхности |
| Surface coverage | q | Поверхностное покрытие |
| Area per molecule in complete monolayer of substance i | am(i) | Площадь на молекулу в заполненном монослое вещества i |
| Surface site | * | Поверхностный центр |
| Ion Мn+ (or atom М) of adsorbent or catalyst at the surface | (или Ms) | Ион Мn+ (или атом М) адсорбента или катализатора на поверхности |
| Constant in Henry's law | K | Константа закона Генри |
| Constant in Langmiur's adsorption isotherms | K | Константа изотермы адсорбции Лэнгмюра |
| Constant in Langmiur's adsorption isotherm for substance i | Ki | Константа изотермы адсорбции Лэнгмюра для вещества i |
| Constants in Freundlich adsorption isotherms | a, n | Константы изотерм адсорбции Фрейнлиха |
| Constants in Temkin isotherms | A, B | Константы изотерм Темкина |
| Constant in BET isotherms | c | Константа изотерм БЭТ |
| Monolayer capacity | nm | Емкость монослоя |
| Constants of Roginskii-Zeldovich equation | a, b | Константы уравнения Рогинского-Зельдовича |
| 2.3 Composition, structure and texture of catalysts | 2.3 Состав, структура и текстура катализаторов | |
| 2.4 Catalytic reactors | 2.4 Каталитические реакторы | |
| 2.5 Kinetics of heterogeneous catalytic reactions | 2.5 Кинетика гетерогенных каталитических реакций | |
| Stoichiometric coefficient of substance B |
nB | Стехиометрический коэффициент вещества B |
| Extent of reaction | x | Степень реакции |
| Rate of catalysed reaction | xm | Скорость катализируемой реакции |
| Quantity of catalyst | Q | Количество катализатора |
| Specific rate of reaction | rm | Удельная скорость реакции |
| Specific activity of the catalyst | rm | Удельная активность катализатора |
| Rate of reaction per unit volume of catalyst | rv | Скорость реакции на единицу объема катализатора |
| Areal rate of reaction | ra | Поверхностная скорость реакции |
| Turnover frequency (turnover number) |
N | Частота оборотов (число оборотов) |
| Selectivity | S, SF, SR | Селективность |
| Rate constant | k | Константа скорости |
| Order of the reaction | ai | Порядок реакции |
| Frequency factor | A | Частотный фактор |
| Activation energy | E | Энергия активации |
| Isokinetic temperature (Kelvin scale) | Tθ | Изокинетическая температура (шкала Кельвина) |
| Fraction converted | x | Степень превращения |
| Feed rate | n | Скорость подачи |
| Space velocities | nm, nn , na | Объемные скорости |
| Space times | tm,tn ,ta | Объемное время |
| Sum of surface concentrations of reaction centres | L | Сумма поверхностных концентраций центров реакции |
| Surface concentration of surface intermediate m | Lm | Поверхностная концентрация поверхностного интермедиата m |
| Surface concentration of vacant reaction centres | Lν | Поверхностная концентрация вакантных центров реакции |
| Energy of activation for activated adsorption | Eads | Энергия активации активированной адсорбции |
| Energy of activation for activated adsorption on uncovered surface |
Энергия активации активированной адсорбции на свободной поверхности | |
| (Differential) enthalpy of adsorption |
- q | (Дифференциальная) энтальпия адсорбции |
| (Differential) enthalpy of adsorption on uncovered surface | - q0 | Дифференциальная) энтальпия адсорбции на свободной поверхности |
| Transfer coefficient | a | Коэффициент переноса |
| 2.6 Transport phenomena in heterogeneous catalysis | 2.6 Транспортные явления в гетерогенном катализе | |
| 2.7 Loss of catalytic activity | 2.7 Потеря каталитической активности | |
| Constants in equation for rate of deactivation | B, n | Константы уравнения скорости дезактивации |
| Time of run (on stream) | t | Время опыта (в работе) |
| 2.8 Mechanism | 2.8 Механизм | |
| 2.9 Nomenclature of catalytic reactions | 2.9 Номенклатура каталитических реакций |
Перевод Е. Никифорово
1.6 Transport phenomena in heterogeneous catalysis
This section will not attempt to cover the more technical aspects of chemical reactor engineering.
A unique feature of heterogeneous catalytic reactions is the ease with which chemical kinetic laws are disguised by various transport phenomena connected with the existence of concentration and/or temperature gradients in the hydrodynamic boundary layer surrounding the catalyst particles (external gradients) or in the porous texture of the catalyst particles themselves (internal gradients). Additional difficulties arise in batch reactors and in stirred flow reactors if agitation is inadequate to maintain uniform concentrations in the fluid phase. Agitation is particularly critical where one of the reactants is a gas and the catalyst and other reactants and products are in condensed phases, for example, in the hydrogenation of a liquid alkene. Here the agitation must be adequate to maintain the fugacity of the dissolved gaseous reactant equal to that in the gaseous phase.
When external gradients correspond to substantial differences in concentration or temperature between the bulk of the fluid and the external surface of the catalyst particle, the rate of reaction at the surface is significantly different from that which would prevail if the concentration or temperature at the surface were equal to that in the bulk of the fluid. The catalytic reaction is then said to be influenced by external mass or heat transfer, respectively, and, when this influence is the dominant one, the rate corresponds to a regime of external mass or heat transfer.
Similarly, when internal gradients correspond to differences in concentration or temperature between the external surface of the catalyst particle and its centre, the rate in the particle is substantially different from that which would prevail if the concentration or temperature were the same throughout the particle. The catalytic reaction is then said to be influenced by internal mass or heat transfer, and, when this influence is the dominant one, the rate corresponds to a regime of internal mass or heat transfer.
Terms such as diffusion limited or diffusion controlled are undesirable because a rate may be larger in regimes of heat or mass transfer than in the kinetic regime of operation, i.e., when gradients are negligible.
1.7 Loss of catalytic activity
1.7.1 Poisoning and inhibition
Traces of impurities in the fluid to which the catalyst is exposed can adsorb at the active sites and reduce or eliminate catalytic activity. This is called poisoning and the effective impurity is called a poison. If adsorption of poison is strong and not readily reversed, the poisoning is called permanent. If the adsorption of the poison is weaker and reversible, removal of the poison from the fluid phase results in restoration of the original catalytic activity. Such poisoning is called temporary. If adsorption of the poison is still weaker and not greatly preferred to adsorption of reactant, the reduction in rate occasioned by the poison may be called competitive inhibition or inhibition. Here, of course, the poison may be present in much larger than trace amounts, There are, of course, no sharp boundaries in the sequence permanent poisoning, temporary poisoning, competitive inhibition.
In selective poisoning or selective inhibition, a poison retards the rate of one catalysed reaction more than that of another or it may retard only one of the reactions. For example, there are poisons which retard the hydrogenation of olefins much more than the hydrogenation of acetylenes or dienes. Also, traces of sulphur compounds appear selectively to inhibit hydrogenolysis of hydrocarbons during catalytic reforming.
A product of a reaction may cause poisoning or inhibition. The phenomenon is called self-poisoning or autopoisoning.
1.7.2 Deactivation: general
The conversion in a catalytic reaction performed under constant conditions of reaction often decreases with time of run or time on stream. This phenomenon is called catalyst deactivation or catalyst decay. If it is possible to determine the kinetic form of the reaction and, thus, to measure the rate constant for the catalytic reaction k, it is sometimes possible to express the rate of deactivation by an empirical equation such as ,where t is the time on stream, n is some positive constant, and B remains constant during a run but depends upon the temperature and other conditions of the reaction. Alternatively, the decline in k may be assumed to result from elimination of active sites and L may be substituted for k in the preceding equation where L is considered to be the effective concentration of surface centres. It is then common practice to define a time of deactivation (or decay time) as the time on stream during which k falls to a specified fraction of its original value, often 0.5. Times of deactivation may vary from minutes as in catalytic cracking to years as in hydrodesulphurisation.
Catalytic deactivation can sometimes be reversed and the original catalytic activity restored by some special operation called regeneration. For example, coked cracking catalyst is regenerated by burning off the coke (see §§1.7.3, 1.9).
If the catalytic reaction is a network of various processes, deactivation can lead to a change in the distribution of products. In such cases, the deactivation not only reduces the overall rate but it changes the selectivity.
1.7.3 Types of deactivation
Catalyst deactivation can result from deactivation of catalytic sites by poisoning either by impurities or by products of the catalytic reaction (§1.7.1). Many reactions involving hydrocarbons and particularly those run at higher temperatures lead to the deposition on the catalyst of high molecular weight compounds of carbon and hydrogen which deactivate the catalyst. This phenomenon is called coking or fouling. Catalysts so deactivated can often be regenerated.
Catalyst deactivation may also result from changes in the structure or in the texture of the catalyst. Changes of this kind are usually irreversible and the catalyst cannot be regenerated. This type of deactivation is often called catalyst ageing.
Sintering and recrystallisation. Catalysts often suffer during use from a gradual increase in the average size of the crystallites or growth of the primary particles. This is usually called sintering. The occurrence of sintering leads to a decrease in surface area and, therefore, to a decrease in the number of catalytic sites. In some cases, sintering leads to a change in the catalytic properties of the sites, for example, for catalysts consisting of highly dispersed metals on supports, catalytic properties may change on sintering due to a change in the relative exposure of different crystal planes of the metallic component of the catalyst or for other reasons. Thus sintering leads to a decrease in rate and perhaps also to a change in selectivity. Similar phenomena can occur in oxide catalysts as used in catalytic oxidation. The crystal size increases, or the initial structure of the crystals changes. For example, a binary solid compound may decompose into its components or an amorphous mass may crystallise. These processes may be called recrystallisation. In some cases the terms sintering and recrystallisation may refer to the same process. The removal of surface defects may accompany these processes.
In some cases, as for example in catalytic cracking on silica-alumina, processes similar to those involved in sintering and recrystallisation can lead to a change in the texture of the catalyst. Surface areas are diminished and the pore-size distribution is changed.
1.8 Mechanism of catalytic reactions
1.8.1 General
A chemical reaction proceeds by a set of elementary processes (the Manual, §11.3) which are in series and perhaps also in parallel. These processes start and terminate at species of minimum free energy (reactants, intermediates and products) and each elementary process passes through a state of maximum free energy (the transition state). To specify the mechanism, one must specify the elementary processes. This specifies the intermediates. One must also give the nature (energetics, structure, charge distribution) of the transition state. So much is true for chemistry in general. The special features of mechanism in heterogeneous catalysis are those which involve reactions between sorptives and active sites, reactions among adsorbates, and processes which regenerate active sites to give a type of chain reaction.
In general, only partial approaches to the specification of mechanism as given above have been possible.
Mechanism is sometimes used in different senses. For example, consider the two situations.
|
A + B C |
vs |
A + B C |
|
C D |
C D |
It may be said that the two situations have different mechanisms or that they are two variants of the same mechanism.
1.8.2 Elementary processes in heterogeneous catalysis
There are many more types of elementary processes in heterogeneous catalysis than in gas phase reactions. In heterogeneous catalysis the elementary processes are broadly classified as either adsorption-desorption or surface reaction, i.e., elementary processes which involve reaction of adsorbed species. Free surface sites and molecules from the fluid phase may or may not participate in surface reaction steps.
There is no generally accepted classification of elementary processes in heterogeneous catalysis. However, names for a few types of elementary processes are generally accepted and terminology for a partial classification [see M. Boudart, Kinetics of Chemical Processes, Chap. 2 (1968)] has received some currency. The particular reactions used below to exemplify this terminology are ones which have been proposed in the literature but some have not been securely established as occurring in nature at any important rate.
Adsorption-desorption. This includes the process of physical adsorption as well as non-dissociative chemisorption.
* + NH3(g) H3N*
* + H(g) H*
Dissociative adsorption and its reverse, associative desorption.
2* + CH4(g) CH3* + H*
The methane might be supposed to react either from the gas phase or from a physisorbed state.
Dissociative surface reaction and its reverse, associative surface reaction.
2* + C2H5* H* + *CH2CH2*
This involves "dissociative adsorption" in an adsorbate.
Sorptive insertion. This is analogous to the process of ligand insertion in coordination chemistry.
H* + C2H4(g) *C2H5
This reaction might also be imagined to proceed by adsorption of C2H4 followed by ligand migration (an associative surface reaction).
Reactive adsorption and its reverse, reactive desorption. This resembles dissociative adsorption but one fragment adds to an adsorbate rather than to a surface site. In abstraction and extraction processes, an adsorptive or adsorbate species extracts an adsorbed atom or a lattice atom, respectively.
Abstraction process *H + H(g) * + H2(g)
Extraction process + CO(g) 2e + CO2(g)
The following elementary process occurring either on one site or, as shown, on two sites is called a Rideal or a Rideal-Eley mechanism:
| D - D(g) | H - D(g) | |
| H | ||
| * * | |
* * |
D2 may also be considered to be in some kind of a weakly adsorbed state. It will be noted that one D atom is never bonded to the surface in any minimum Gibbs energy intermediate. It is recommended that the term Rideal or Rideal-Eley mechanism be reserved for this particular elementary process. However, the term has been used for analogous processes in which there is a reactant molecule and a product molecule of nearly the same energy in the fluid phase or in some weakly adsorbed state and in which one or more atoms are never bonded to the surface. An example is the following elementary process, which has been called a switch process. The term might well be used generically for similar processes. The term Rideal or Rideal-Eley mechanism has been further extended to include all elementary processes in which a molecule reacts from the fluid phase or from some weakly adsorbed state. Even the sorptive insertion process and the abstraction process illustrated above fall within this extended definition.
1.8.3 Nomenclature of surface intermediates
Surface intermediates should be named in ways compatible insofar as possible with chemical nomenclature in general.
Adsorbed species may be treated as surface compounds analogous to molecular compounds. For example, *H may be called surface hydride, *=C=O may be called a linear surface carbonyl and may be called a bridged surface carbonyl. H2N* may be called a surface amide and H3C* a surface methyl or a surface s -alkyl.
The species *H may also be called an adsorbed hydrogen atom and *CO, adsorbed carbon monoxide.
Organic adsorbates pose a particular problem because quite particular structures of some complexity are regularly discussed. A nomenclature is recommended in which the surface is treated as a substituent which replaces one or more hydrogen atoms. The degree of substitution is indicated by monoadsorbed, diadsorbed, etc. This terminology does not specify the nature of the chemical bonding to the surface nor does it restrict, a priori, the valency of the surface site *. Thus, both of the following species are named 1,3-di-adsorbed propane. Other examples are:
| *CH3 | monoadsorbed methane |
| *CH2CH2CH3 | 1-monoadsorbed propane |
| 2-monoadsorbed 2-methylpropane |
|
| *OCH2CH3 | O-monoadsorbed ethanol |
| *CH2CH2OH | 2-monoadsorbed ethanol |
| * = CH - CH3 or (*)2CH-CH3 | 1,1-diadsorbed ethane |
| * = NH or (*)2NH | diadsorbed ammonia |
| *COCH3 | 1-monoadsorbed acetaldehyde |
| Species adsorbed as p-complexes are described as p -adsorbed: | |
| p-adsorbed allyl | |
The substitution system of nomenclature should be viewed as showing only how atoms are connected and not as indicating the precise electronic structure. Thus p -adsorbed ethylene is one representation of 1,2-diadsorbed ethane.
Nomenclature based upon the process of formation of a particular adsorbate is to be discouraged. Thus, H* may be "dissociatively adsorbed hydrogen" but the same species is formed in dissociative adsorption of CH4, NH3, H2O.
1.9 Nomenclature of catalytic reactions
In general, a catalytic reaction may be named by adding the adjective "catalytic" to the standard chemical term for the reaction, for example, catalytic hydrogenation (or, if clarity demands, heterogeneous catalytic hydrogenation), catalytic hydrodesulphurisation, catalytic oxidative dehydrogenation, catalytic stereospecific polymerisation.
In general, special terminology for reactions is to be discouraged. However, certain catalytic processes of technological interest have special names in common use. Where such processes involve the simultaneous occurrence of two or more different chemical reactions, special names for the processes are probably inevitable. Some Important examples of such processes of technological interest are:
Catalytic cracking. In this process, a higher boiling cut of petroleum, for example, gas oil, is converted substantially into a lower boiling material of high octane number. Among the processes which appear to
be involved are skeletal isomerisation of alkanes followed by their cleavage into alkane and olefin, and hydrogen transfer reactions which reduce the amount of olefin formed and which lead to coke and aromatic hydrocarbons.
Catalytic hydrocracking. This is similar to catalytic cracking in its industrial purpose but it is effected under hydrogen pressure and on a catalyst containing an ingredient with a hydrogenating function.
Catalytic reforming. Catalytic reforming is a process for increasing the octane number of naphthas. It involves isomerisation of alkanes, dehydrogenation of cyclohexanes to aromatic hydrocarbons, isomerisation and dehydrogenation of alkylcyclopentanes, and dehydrocyclisation of alkanes.
The following reactions may be mentioned because they are rare except as heterogeneous catalytic reactions and have somewhat specialised meanings in catalysis.
Catalytic methanation. This is a process for removing carbon monoxide from gas streams or for producing methane by the reaction
CO + 3H2 CH4 + H2O
Catalytic dehydrocyclisation. This is a reaction in which an alkane is converted into an aromatic hydrocarbon and hydrogen, for example,
heptane toluene + 4H2
Catalytic hydrogenolysis. This is ordinarily used for reactions in which º C-Cº + H2 gives º CH + HCº , for example,
propane + H2 ethane + methane
toluene + H2 benzene + methane
butane + H2 2 ethane
However, it may also be used for cleavage of bonds other than º C-Cº , for example,
benzyl acetate + H2 toluene + acetic acid
benzylamine + H2 toluene + NH3
Catalytic hydrodesulphurisation. This is a process in which, in the presence of hydrogen, sulphur is removed as hydrogen sulphide.
| 2.1 Catalysis and catalysts | 2.1 Катализ и катализаторы | |
| 2.2 Adsorption | 2.2 Адсорбция | |
| Area of surface | A, As, S | Площадь поверхности |
| Specific surface area | a, as, s | Удельная площадь поверхности |
| Surface coverage | q | Поверхностное покрытие |
| Area per molecule in complete monolayer of substance i | am(i) | Площадь на молекулу в заполненном монослое вещества i |
| Surface site | * | Поверхностный центр |
| Ion Мn+ (or atom М) of adsorbent or catalyst at the surface | (или Ms) | Ион Мn+ (или атом М) адсорбента или катализатора на поверхности |
| Constant in Henry's law | K | Константа закона Генри |
| Constant in Langmiur's adsorption isotherms | K | Константа изотермы адсорбции Лэнгмюра |
| Constant in Langmiur's adsorption isotherm for substance i | Ki | Константа изотермы адсорбции Лэнгмюра для вещества i |
| Constants in Freundlich adsorption isotherms | a, n | Константы изотерм адсорбции Фрейнлиха |
| Constants in Temkin isotherms | A, B | Константы изотерм Темкина |
| Constant in BET isotherms | c | Константа изотерм БЭТ |
| Monolayer capacity | nm | Емкость монослоя |
| Constants of Roginskii-Zeldovich equation | a, b | Константы уравнения Рогинского-Зельдовича |
| 2.3 Composition, structure and texture of catalysts | 2.3 Состав, структура и текстура катализаторов | |
| 2.4 Catalytic reactors | 2.4 Каталитические реакторы | |
| 2.5 Kinetics of heterogeneous catalytic reactions | 2.5 Кинетика гетерогенных каталитических реакций | |
| Stoichiometric coefficient of substance B |
nB | Стехиометрический коэффициент вещества B |
| Extent of reaction | x | Степень реакции |
| Rate of catalysed reaction | xm | Скорость катализируемой реакции |
| Quantity of catalyst | Q | Количество катализатора |
| Specific rate of reaction | rm | Удельная скорость реакции |
| Specific activity of the catalyst | rm | Удельная активность катализатора |
| Rate of reaction per unit volume of catalyst | rv | Скорость реакции на единицу объема катализатора |
| Areal rate of reaction | ra | Поверхностная скорость реакции |
| Turnover frequency (turnover number) |
N | Частота оборотов (число оборотов) |
| Selectivity | S, SF, SR | Селективность |
| Rate constant | k | Константа скорости |
| Order of the reaction | ai | Порядок реакции |
| Frequency factor | A | Частотный фактор |
| Activation energy | E | Энергия активации |
| Isokinetic temperature (Kelvin scale) | Tθ | Изокинетическая температура (шкала Кельвина) |
| Fraction converted | x | Степень превращения |
| Feed rate | n | Скорость подачи |
| Space velocities | nm, nn , na | Объемные скорости |
| Space times | tm,tn ,ta | Объемное время |
| Sum of surface concentrations of reaction centres | L | Сумма поверхностных концентраций центров реакции |
| Surface concentration of surface intermediate m | Lm | Поверхностная концентрация поверхностного интермедиата m |
| Surface concentration of vacant reaction centres | Lν | Поверхностная концентрация вакантных центров реакции |
| Energy of activation for activated adsorption | Eads | Энергия активации активированной адсорбции |
| Energy of activation for activated adsorption on uncovered surface |
Энергия активации активированной адсорбции на свободной поверхности | |
| (Differential) enthalpy of adsorption |
- q | (Дифференциальная) энтальпия адсорбции |
| (Differential) enthalpy of adsorption on uncovered surface | - q0 | Дифференциальная) энтальпия адсорбции на свободной поверхности |
| Transfer coefficient | a | Коэффициент переноса |
| 2.6 Transport phenomena in heterogeneous catalysis | 2.6 Транспортные явления в гетерогенном катализе | |
| 2.7 Loss of catalytic activity | 2.7 Потеря каталитической активности | |
| Constants in equation for rate of deactivation | B, n | Константы уравнения скорости дезактивации |
| Time of run (on stream) | t | Время опыта (в работе) |
| 2.8 Mechanism | 2.8 Механизм | |
| 2.9 Nomenclature of catalytic reactions | 2.9 Номенклатура каталитических реакций |
Перевод Е. Никифорово
15 - 17 июня 2005 года г. Омск
Доктор химических наук, профессор ЮРИЙ ИВАНОВИЧ ЕРМАКОВ - инициатор становления нового направления в катализе, связанного с созданием научных основ приготовления нанесенных катализаторов путем целенаправленного синтеза поверхностных соединений. Созданная им научная школа продолжает исследования в области конструирования нанесенных катализаторов, установления механизма их действия на молекулярном уровне и создания новых высокоэффективных катализаторов.
Ю.И. Ермаков внес существенный вклад в создание в 1978 г. Омского отдела каталитических превращений углеводородов, выросшего впоследствии в Институт проблем переработки углеводородов СО РАН.

Конференция "Молекулярный дизайн катализаторов и катализ в процессах переработки углеводородов и полимеризации", приуроченная к 70-летию со Дня рождения профессора Ю.И. Ермакова, прошла с участием ученых из стран СНГ (Казахстан) и стран дальнего зарубежья (Бразилия, Венгрия, США).
В работе конференции приняли участие 89 человек, в том числе:
На конференции было заслушано 6 ключевых лекций, 30 устных и представлен 21 стендовый доклад по следующим секциям:
Заместитель председателя Оргкомитета д.х.н. В.А. Захаров выступил с сообщением о жизни и деятельности профессора Ю.И. Ермакова.
Научная часть конференции открылась ключевой лекцией Prof. Alexis T. Bell (Department of Chemical Engineering University of California, Berkeley, USA) "Single Site Catalysts - Ideal Models for Understanding Structure-Function Relationships". В докладе были освещены пути преодоления трудностей при проведении исследований активных центров и каталитических свойств катализаторов, представляющих собой наноразмерные металлические частицы, а также оксиды или сульфиды металлов, диспергированные на поверхности носителя, переведением их в формы изолированных катионов или оксометаллические группы.
В лекции чл.-корр. РАН В.А. Лихолобова (Институт проблем переработки углеводородов СО РАН, г. Омск) были рассмотрены тенденции развития исследований в области молекулярного дизайна активных центров катализаторов. Отмечено, что актуальными направлениями в этой области в настоящее время являются гомогенизация гетерогенных систем (гигантские кластеры, стабилизированные коллоиды, дендримеры) и сочетание гетерогенизированных гомогенных систем с электро-, фото- и биокатализаторами.
Изучению механизмов закрепления металлокомплексов на носителях как подхода к формированию молекулярной структуры предшественников активного компонента в катализаторах превращения углеводородов была посвящена ключевая лекция д.х.н.
В.К. Дуплякина (Институт проблем переработки углеводородов СО РАН, г. Омск).
Д.х.н. Г.В. Ечевский (Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск) посвятил свою лекцию нетрадиционной одностадийной технологии получения высококачественных моторных топлив из широкой фракции углеводородов (нк - 360 oС) - БМИТ. Докладчик отметил преимущественные особенности катализатора, применяемого данной технологией. Было отмечено, что гибкость технологии позволяет при смене сырья сохранять номенклатуру и качество получаемых топлив.
В лекции о роли носителей в формировании активного компонента нанесенных катализаторов полимеризации олефинов д.х.н. В.А. Захаров (Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск) обратил внимание слушателей на то, что основой для разработки и исследования большого числа новых нанесенных катализаторов полимеризации различного состава послужили пионерские работы Ю.И. Ермакова, выполненные в конце 60-х - начале 70-х г.г. прошлого века. В настоящем сообщении докладчик, основываясь на известных литературных данных и новых экспериментальных результатах, рассмотрел схемы формирования активного компонента нанесенных катализаторов полимеризации различного состава, включая традиционные окиснохромовые катализаторы и наиболее новые системы на основе металлоценовых и постметаллоценовых комплексов переходных металлов. В заключении был сделан вывод о том, что в зависимости от состава катализатора реализуются различные схемы формирования активного компонента каталитической системы. В ряде случаев природа носителя может существенно влиять на реакционную способность образующихся активных центров.
Òема ключевой лекции к.х.н. Н.М. Бравой (Институт прîáлем химической физики РАН, Черноголовка, Московская оáëасть) - "Триизобутилалюминий и основания Льюиса как мîäификаторы свойств гомогенных металлоценовых катализатîðов полимеризации олефинов". Докладчик отметила актуалüíость данной работы, привела привлекательные характеристèêи и факторы, сдерживающие практическое применение метаëëоценовых катализаторов, в частности, отставание в разработке эффективных иммобилизованных металлоценовых катализатîðов и необходимость использования дорогих и непрактичных активаторов. Н.М. Бравая представила в докладе примеры ряда новых путей активации гомогенных металлоценовых катализаторов с использованием комбинированного активатора, МАО/триизобутилалюминий (ТИБА), рассмотрела вопросы, связанные со степенью участия ТИБА в активации и модификации стереоселективности гомогенной металлоценовой каталитической системы в ходе полимеризации пропилена. Показала возможность модификации каталитических свойств (активность, стереоселективность) гомогенных металлоценовых систем под действием оснований Льюиса.
На конференции работало три секции:
1. Секция "Молекулярный дизайн в приготовлении и исследовании нанесенных катализаторов".
Было заслушано 11 устных докладов, в том числе: Borbath I., (Institute of Surface Chemistry and Catalysis, Hungarian Academy of Sciences, Budapest, Hungary) об использовании метода поверхностной органометаллической химии для получения активных центров катализаторов, представляющих собой оловосодержащие соединения платиновых металлов на поверхности оксидов кремния и алюминия; Prof. Gusevskaya E.V. (Universidade Federal de Minas Gerais, Belo Horizonte, Brasil) об окислительных превращениях монотерпенов на палладий- и кобальтсодержащих катализаторах.
О новом подходе к формированию активного центра катализатора через поверхностный синтез полигидроксокомплексов платины рассказала Бельская О.Б. (ИППУ СО РАН, Омск), об использовании гидроксокомплексов для синтеза предшественников алюмоплатиновых катализаторов - д.х.н. Лисицын А.С. (ИППУ СО РАН), о молекулярном дизайне и исследовании катализаторов крекинга нового поколения - к.х.н. В.А. Дроздов (ИППУ СО РАН), о разработке катализаторов с низким содержанием платины для катодов ТПТЭ - д.х.н. Исмагилов З.Р. (ИК СО РАН, Новосибирск). Д.х.н. Кузнецов Б.Н. (ИХХТ СО РАН, Красноярск) представил доклад о результатах, достигнутых при изучении формирования наноразмерных частиц палладия на углеродных подложках из природных графитов и антрацитов. В докладе д.х.н. В.Ф. Третьякова (ИНХС им. А.В. Топчиева РАН, Москва) представлены результаты ИК-спектроскопических исследований свойств поверхностных соединений в превращении метанола. К.х.н. Максимов Г.М. (ИК СО РАН, Новосибирск) доложил о синтезе и исследованиях гетерогенных кислотных W-Mo катализаторах, которые эффективно катализируют жидкофазные реакции (в частности, разложения гидроперекиси кумола и синтеза витамина Е) и способны заменить в них минеральные кислоты.
Успешно выступили и молодые научные сотрудники:
Завьялова У.Ф. (ИНХС им. А.В. Топчиева РАН, Москва) с сообщением на тему "Разработка и исследование нанесенных CuCo2Ox катализаторов глубокого окисления углеводородов, синтезированных методом горения", Ищенко А.В (ИК СО РАН, Новосибирск) доложил о результатах исследования методом электронной микроскопии высокого разрешения распределения меди в дефектной структуре оксида цинка в Cu/ZnO катализаторах синтеза метанола.
2. Секция "Катализ в процессах переработки углеводородного сырья".
Заслушано 14 устных докладов. Открыл работу секции доклад д.х.н. В.Г. Степанова (Научно-инженерный центр "Цеосит" ОИК СО РАН, Новосибирск), в котором рассмотрены вопросы, касающиеся производства дизельного топлива и автобензинов на минизаводах удаленных регионов, обладающих запасами углеводородного сырья. Докладчик привел результаты сравнительного анализа переработки прямогонных бензинов различными вторичными процессами и в заключение сделал вывод о том, что для производства автобензинов на малотоннажных установках наиболее пригоден процесс "цеоформинг". Вопросам получения жидких углеводородных моторных топлив из этанола на цеолитных катализаторах был посвящен доклад д.х.н. В.Ф. Третьякова (ИНХС им. А.В. Топчиева РАН, Москва), в котором показано, что в зависимости от условий проведения реакции возможно получение ряда ценных органических продуктов, от этилена и лёгких олефинов до углеводородов, входящих в состав моторного топлива. К.х.н. Абрамова А.В. (ИНХС им. А.В. Топчиева РАН, Москва) посвятила свой доклад проблемам изомеризации углеводородов бензиновой фракции синтеза Фишера-Тропша на цеолитсодержащих катализаторах, Делий И.В. (ИК СО РАН, Новосибирск) - процессу жидкофазной изомеризация b -пинена на металлах платиновой группы. В своем докладе Кихтянин О.В. (ИК СО РАН, Новосибирск) продемонстрировал, что катализаторы на основе систем Pt-SAPO-31, являются высокоэффективными катализаторами гидропревращения различных фракций углеводородов, в том числе и с большой молекулярной массой.
Результаты, полученные при конструировании новых модификаций цеолитсодержащих катализаторов крекинга, представили в своих докладах сотрудники ИППУ СО РАН, г.Омск: к.т.н.
Доронин В.П., к.х.н. Чжу Д.П., Белая Л.А. В докладе к.х.н.
Кадирбекова К.А. (Институт химических наук им. А.Б. Бектурова МОН РК, Алматы, Казахстан) показана возможность использования модифицированных природных цеолитов Шанканайского месторождения (Казахстан) в качестве катализаторов для крекинга технического парафина в альфа-олефины. На катализаторах, полученных на основе модифицированного природного цеолита, максимальная конверсия парафина составляет 56,0 % при выходе целевого продукта 31,9 %. Д.х.н. Старцев А.Н. (ИК СО РАН, Новосибирск) в докладе "Низкотемпературное разложение сероводорода" привел экспериментальные свидетельства возможности осуществления реакции процесса при низких температурах со 100 % конверсией и селективностью и представил энергетический профиль данной реакции. Большой интерес вызвали доклады к.х.н. Данилюка А.Ф. "Мезопористые катализаторы и носители на основе кремнезема и углерода" (ИК СО РАН, Новосибирск), Данилова А.В. "гидропереработка нефтяных фракций на полифункциональных катализаторах" (ИОКЭ им. Д.В. Сокольского, МОН РК, Алматы, Казахстан). В работе к.х.н. Шилиной М.И. (МГУ им. М.В. Ломоносова, Москва) на примере конверсии парафинов изучены каталитические свойства промотированного хлорида алюминия в условиях ограниченной молекулярной подвижности при низких температурах. Показано, что механизм с участием неклассических карбониевых ионов, рассматриваемый обычно для реакций углеводородов в протонных суперкислотных средах, может реализовываться и в матрицах углеводородов под действием апротонных кислот Льюиса, в частности, промотированного солями кобальта хлорида алюминия. В докладе д.х.н. Белого А.С. (ИППУ СО РАН, Омск) сообщалось об исследовании эффектов модифицирования оксида алюминия элементами VI группы на адсорбционные и каталитические свойства платины в катализаторах риформинга и представлены результаты использования этих эффектов в производстве промышленных катализаторов.
3. Секция "Каталитическая полимеризация олефинов (новые каталитические системы, состав и строение катализаторов, кинетика полимеризации и структура полимеров)".
Представлено 6 устных докладов. Секция начала работу с доклада к.х.н. Т.Б. Микенас (ИК СО РАН, Новосибирск), посвященного исследованию влияния состава новых нанесенных титан- и ванадиймагниевых катализаторов на молекулярные характеристики полиэтилена. В сообщении к.х.н. Букатова Г.Д. (ИК СО РАН, Новосибирск) на тему "Число и реакционная способность активных центров при полимеризации олефинов на нанесенных титанмагниевых катализаторах" представлены новые данные по определению числа активных центров (СР) и константы скорости роста (КР) при полимеризации этилена и пропилена на высокоактивных титанмагниевых катализаторах (ТМК) разного состава методом обрыва полимеризации радиоактивной окисью углерода.
Применение высокоэффективных гомогенных металлоценовых катализаторов для направленного регулирования микроструктуры полимерной цепи показано в работах, представленных к.х.н. Недорезовой П.М. (ИХФ им. Н.Н. Семенова РАН, Москва) и А.Н. Клямкиной (ИХФ им. Н.Н. Семенова РАН, Москва). В этих докладах исследован процесс полимеризации и сополимеризации пропилена с рядом со-мономеров в среде жидкого пропилена с использованием гомогенных высокоактивных каталитических систем на основе широкого ряда анса-металлоценов С1, C2 ,Сs-симметрии, изучена взаимосвязь между составом и структурой металлоценов и их активностью, сополимеризующей способностью и свойствами образующегося ПП. Показана возможность получения образцов ПП с широким диапазоном свойств варьирования структуры анса-металлоценов.
В докладе к.х.н. Дубкова К.А. (ИК СО РАН, Новосибирск) рассмотрен новый метод введения карбонильных групп в полимеры, содержащие двойные С=С связи, основанный на реакции карбоксидирования, которая осуществляется путем некаталитического взаимодействия закиси азота с С=С связями полимера. Описанный метод позволяет проводить химическую модификацию полимеров, а также получать функционализированные олигомеры с заданным содержанием С=О групп и регулируемой молекулярной массой.

Таким образом, во время работы конференции были представлены результаты многих интересных научных работ и были обсуждены современные подходы к решению проблем, касающихся как научных основ приготовления и молекулярного дизайна катализаторов, так и применения катализа в процессах переработки углеводородов и полимеризации.
Подводя итоги конференции, участники единодушно отметили, что научное направление в катализе, связанное с созданием научных основ приготовления нанесенных катализаторов путем целенаправленного синтеза поверхностных соединений, инициатором которого был д.х.н. Ю.И. Ермаков, продолжает развиваться не только в научных коллективах России, но и за рубежом. Научная школа профессора Юрия Ивановича Ермакова успешно работает, сохраняя преемственность и совершенствуя заложенные им научные направления.
Секретариат конференции
Фото А. Слептерева
26-29 июня 2005 года в Доме ученых СО РАН (Новосибирск) прошло Международное рабочее совещание "Происхождение и эволюция биосферы". Совещание было призвано собрать специалистов разных направлений, чтобы обеспечить комплексный подход к проблеме происхождения и эволюции биосферы. В совещании приняли участие 200 человек, в том числе 12 иностранных ученых из 5 стран. Российские участники были представлены 68 организациями.
На совещании было заслушано 24 пленарные лекции, 29 устных докладов и представлено 80 стендовых докладов по следующим направлениям:
Проведены Круглые столы по укрупненным направлениям. Полные тексты докладов будут опубликованы в журнале Biogeosciences. Организаторами совещания являлись Объединенный институт геологии, геофизики и минералогии им. А.А. Трофимука СО РАН (Новосибирск), Институт катализа им. Г.К. Борескова СО РАН (Новосибирск), Институт цитологии и генетики СО РАН (Новосибирск), Палеонтологический институт РАН (Москва). Необходимо отметить участие в совещании 19 действительных членов Российской Академии наук, что несомненно свидетельствует о его высоком научном уровне.
Тематика пленарных лекций отразила 8 основных направлений программы президиума РАН, которая легла в основу научной проблематики совещания. Три основополагающие лекции обеспечили возможность комплексного подхода к обсуждению проблем происхождения и эволюции биосферы. Академик Н.Л. Добрецов (Объединенный институт геологии, геофизики и минералогии
им. А.А. Трофимука СО РАН, Новосибирск) в своей лекции "О ранних стадиях зарождения и эволюции жизни" констатировал, что эволюция жизни началась примерно 3,8 млрд лет назад с "Мира РНК". Базой мог быть абиогенный синтез олигонуклеотидов РНК на монтмориллоните. Появление путем спонтанной рекомбинации и обмена олигонуклеотидами длинных макромолекул РНК с рибозимной активностью замкнуло цикл матричного воспроизводства РНК. Эксперименты, а также тот факт, что все базовые процессы в живой клетке могут идти лишь при помощи имеющихся в ней РНК разных типов, поддерживают такой сценарий. Возникнув примерно
3,6 млрд лет назад, триада "ДНК-РНК-белок" и липидная мембрана сформировали живые клетки. Условия ранней Земли заставляют предположить, что это могли быть клетки архебактерий. Огромное разнообразие прокариот в сочетании с консерватизмом прокариотных экосистем заставляют предполагать особые принципы эволюции в "Мире прокариот": горизонтальный перенос генов, формирующий сетевую картину эволюции и адаптивная динамика сообществ симбионтов-синтрофов, причем наиболее стойкие симбиотические связи наблюдаются как раз между таксономически далекими прокариотами. Ярким примером таких сообществ служат цианобактериальные маты, практически не изменившиеся за 3,6 млрд лет.
Академик А.С. Спирин (Институт белка РАН, Пущино) в своей лекции "Мир РНК и его эволюция" рассмотрел идею первичности РНК в происхождении жизни на Земле и гипотетический древний мир РНК. Основываясь на современных представленных о много-функциональности (омнипотентности) РНК, автор рассматривает три новых механизма, участие которых в проиñõождении и эволюции древнего мира РНК могли играть критèческую роль: (1) реакция спонтанной трансэстерификации полирибонуклеотидов, открытая А.Б. Четвериным с сотрудникамии, которая могла приводить к удлинению первичных коротких олигорибонуклеотидов и к генерации вариантов последовательностей для последующего отбора; (2) компартментализация функциональных ансамблей РНК в виде смешанных молекулярных колоний на влажных твердых средах (глинах), в отсутствие оболочек и мембран; (3) систематическое экспоненциалüíое обогащение популяции РНК функционально лучшими молекулами за счет попеременного растворения колоний при затоплении и образования новых колоний при подсушивании первобытных водоемов ("первобытный естественный SELEX"). Предполагается, что первыми живыми особями (организмами) на Земле могли быть временные колонии самовоспроизводящихся ансамблей молекул РНК на влажных минеральных средах.
С большим вниманием аудиторией была заслушана лекция академика Г.А. Заварзина (Институт микробиологии им. С.Н. Виноградского РАН, Москва) "Становление биосферы". Становление биосферы начинается с появления первых организмов. Обитаемость предшествует обитанию. Поэтому приоритет принадлежит определяющим геосферным условиям. Основу взаимодействия геосферы и биосферы составляет система сîïряженных биогеохимических циклов с ведущим циклом органического углерода. Становление биосферы происходило за счет деятельности прокариотных организмов. Устойчивое развитие (эволюция) биосферы требует функционального разнообразия и не может быть сведено к универсальному общему предку. Поскольку все новое для своего утверждения должно согласовываться с уже существующим, то есть старым, то эволюция должна вписываться в рамки прокариотной биосферы. В этом смысле эволюция аддитивна, а не субституитивна. Замена происходит внутри функциональной ниши в устойчивой системе. Цикл органического углерода определяется первичными продуцентами. Поэтому эволюция биосферы есть в первую очередь эволюция фотосинтезирующих организмов, зависящих от Солнца, и отчасти от хемосинтезирующих организмов, зависящих от эндогенных окислительно-восстановительных реакций.
Направление "Абиогенный синтез и химическая эволюция вещества на догеологических этапах формирования Земли" было представлено лекциями к.ф.-м.н. В.Н. Снытникова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) "Астрокатализ: происхождение органических предбиологических соединений на земле", в которой нашла отражение идея "астрокатализа" о происхождении первичного органического вещества, ставшего основой для всех живых организмов на Земле (и сегодня можно утверждать, что катализ в органическом синтезе сформировал планеты); д.ф.-м.н. В.А. Аветисова (Институт химической физики им. Н.Н. Семенова РАН, Москва) "О современном состоянии проблемы возникновения биологической гомохиральности" и доктора Ричарда Хувера (НАСА, США) "Кометы, углеродсодержащие метеориты и происхождение биосферы".
Вопросы биоминералогии, безматричного синтеза органических соединений на биоминеральных системах отразились в лекциях академика Н.П. Юшкина Институт геологии Коми НЦ УрО РАН, Сыктывкар) "Минеральные предшественники биосистем и концепция углеводородного организмобиоза" и член-корр. РАН С.Л. Вотякова (Институт геологии и геохимии УрО РАН, Екатеринбург) "Структурные дефекты в биокарбонатах как экоиндикаторы и трассеры эволюции геобиологических систем".
Тематика "Мир РНК" была представлена докладом доктора Александра Власова (Somagenics, Inc., США), предлагающего новые представления о раннем становлении мира РНК. Секция "Архейско-протерозойские биологические системы" была представлена четырьмя пленарными докладами - профессора Виктора Мележика (Геологическое общество Норвегии), член-корр. РАН А.Ю. Розанова (Палеонтологический институт РАН, Москва) и профессора Зигфрида Фрэнка (Институт климата, Потсдам, Германия).
Интересные лекции прозвучали в секции "Экосистемно-биоценотическая организация и эволюция", их прочитали член-корр. РАН А.В. Каныгин (Институт геологии нефти и газа СО РАН, Новосибирск) - "Экологические закономерности эволюции биосферы: взаимосвязь кардинальных инноваций в живых системах с геологическими изменениями среды", д.г-м.н. А.С. Алексеев (Палеонтологический институт РАН, Москва) - "Глобальные колебания уровня моря в фанерозое и их движущая роль в экологической и эволюционной динамике морской биоты" и д.б.н. А.Г. Пономаренко (Палеонтологический институт РАН, Москва) - "Изменения наземной биоты в преддверии пермотриасового экологического кризиса".
На секции "Генетические механизмы биологической эволюции и корреляция био-геологических событий" с пленарными лекциями выступили академик С.Г. Инге-Вечтомов (Санкт-Петербургский государственный университет), д.б.н. П.М. Бородин (Институт цитологии и генетики СО РАН, Новосибирск) и академик С.В. Шестаков (Институт общей генетики им. Н.И. Вавилова РАН, Москва). Их лекции отразили вклад генетиков в изучение эволюционных процессов.
Заключала конференцию секция "Механизмы антропогенеза и расселение человека". Лекция д.б.н. А.К. Агаджаняна (Палеонтологический институт РАН, Москва) была посвящена вопросам реконструкции палеосреды и условий обитания древнего человека на примере Северо-Западного Алтая, лекция д.и.н. М.В. Шунькова (Институт археологии и этнографии СО РАН, Новосибирск) -началу верхнего палеолита в Северной Азии.
Устные доклады, прозвучавшие в секциях, дополняли и часто конкретизировали представленный в пленарных лекциях материал. Круглые столы по укрупненным направлениям, а также стендовая сессия продемонстрировали большой интерес участников к проблеме эволюции биосферы. Оживленные дискуссии позволили в непосредственном общении обменяться самыми свежими данными. Необходимо отметить активное участие молодых ученых в прошедшей конференции.
На заключительном заседании конференции сопредседателями Программного комитета академиками Н.Л. Добрецовым и Г.А. Заварзиным было отмечено, что встреча такого формата и такого масштаба по данной проблематике в России проводится впервые. Основная задача - междисциплинарность и комплексный подход к проблеме - была достигнута. От участников поступили предложения о продолжении проведения встреч подобного содержания. Организаторы и участники конференции выразили признательность Российскому фонду фундаментальных исследований за поддержку, которая позволила молодым сотрудникам представить свои доклады и иметь возможность прослушать лекции ведущих ученых, а также принимать участие в дискуссиях.
Т.В. Замулина
Фото А.А. Спиридонова
25-29 июля на турбазе СТИК-ТРЕВЕЛ в Горном Алтае состоялась 2-я Международная Школа-конференция молодых ученых по катализу "Каталитический дизайн - от исследований на молекулярном уровне к практической реализации".
Организаторами конференции выступили Институт катализа им. Г.К. Борескова СО РАН, Новосибирский государственный университет, Российское химическое общество им. Д.И. Менделеева (Новосибирское отделение), Совет Молодых учёных ИК СО РАН, Научный совет по катализу ОХНМ РАН.
Научные направления конференции:
- Катализ в водородной энергетике и электрокатализ;
- Катализ в нефтехимии и нефтепереработке;
- Научные основы приготовления катализаторов, катализ в тонком органическом синтезе;
- Механизмы гетерогенного катализа, методы исследования катализаторов;
- Кинетика и моделирование каталитических реак-ий и реакторов;
- Катализ в защите окружающей среды;
- Правовая охрана и оценка объектов интеллектуальной собственности, поиск информации по научным и коммерческим базам данных, способы трансферта технологий.
В конференции приняли участие 170 человек из 6 стран (Россия, Германия, Польша, Нидерланды, США, Норвегия).
С большим вниманием молодыми учеными были заслушаны 20 лекций новосибирских профессоров и научных сотрудников. Две лекции были представлены московскими учеными: Е.А. Трусовой (Институт нефтехимического синтеза РАН) и Н.В. Круковской (Институт органической химии РАН); одна - доктором Джакобом Вагнером из Института Фрица-Хабера из Берлина (Германия).
Сотрудниками Института катализа было проведено два практикума - "Компьютерный курс по расчету процссов в неподвижном слое катализатора" и "Стратегия использования интеллектуальной собственности в коммерческой деятельности". Молодыми учеными было представлено 37 устных и 62 стендовых доклада. Предварительно были объявлены конкурсы на лучшие доклады для опубликования их полных текстов в виде статей в журналах "Кинетика и катализ" и "Катализ в промышленности". Итоги конкурса были объявлены на закрытии конферен-ии. Новосибирское отделение Российского химического общества им. Д.И. Менделеева учредило премии на два лучших доклада молодых ученых, научное содержание которых соответствовало бы современной и логически выдержанной форме представления и подачи материала. Победителями стали Цёлия Горбатенко (Уральский государственный лесотехнический университет, Екатеринбург) и Сергей Арзуманов (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск).
Основной цлью лекций было привлечь внимание молодых ученых к наиболее важным вопросам, требующим свежего взгляда и оригинальных решений.
Лекция д.х.н. З.Р. Исмагилова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) была посвящена микрореакторам. Ёто направление в последние десять лет активно развивается и сулит большие перспективы. Аппараты малого объема, почти не уступающие большим ни по мощности, ни по набору решаемых задач, имеют модульную конструкцию, что позволяет манипулировать свойствами и "собирать" реактор под конкретные условия и ситуации. Микрореактор - пример реализации идей каталитического молекулярного дизайна. Реакция в нем идет в контролируемых условиях, с заданным теплопереносом, тепловым профилем по реактору. Притом, реакцию можно направленно провести с точностью до одного-двух градусов.
Лекция к.х.н. П.В. Снытникова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) была посвящена проблемам водородной энергетики. Внимание участников школы привлекла лекция к.х.н. И.Г. Даниловой (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) по применению ИК-спектроскопии для охарактеризования свойств поверхности катализаторов и исследования превращения молекул реагентов на активных центрах. Возможности метода позволяют исследовать процессы на молекулярном уровне, начиная со стадии УмокрогоФ синтеза гетерогенных катализаторов, до изучения химии превращений и диффузии молекул реагентов в условиях, максимально приближенных к реализуемым на практике. Близким проблемам была посвящена лекция доктора Джакоба Вагнера из Института Фрица-Хабера, Берлин (Германия).
Развитие научных основ приготовления твердофазных катализаторов - одну из актуальных на сегодняшний день проблем теории и практики катализа - рассмотрел в своей лек-ии к.х.н. Н.А. Пахомов (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск). Большое внимание привлекла лекция д.х.н. В.Б. Фенелонова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск), спе-иалиста в области -еленаправленного формирования внутренней структуры мезопористых материалов. Сейчас синтезируются такие структуры, у которых развивается большая площадь поверхности при малом объеме и мельчайших порах, вполне, однако, достаточных для того, чтобы ввести в них части-ы катализатора. Работают исследователи и над тем, чтобы превратить пассивный носитель катализатора в активного участника про-есса. В.Б. Фенелонов рассказал о получении материалов с высокой площадью поверхности и заданными размерами каналов.
В лекции к.х.н. Е.А. Трусовой (Институт нефтехимического синтеза им. А.В.Топчиева РАН, Москва) были отражены нанотехнологические подходы к проблеме получения новых материалов с заданными свойствами. На современном этапе развития нанотехнологий очень важным является создание теоретической базы в области синтеза и исследования свойств вновь синтезируемых наносистем. Для катализа нанообъекты представляют особый интерес, касается ли это пористой структуры, или размеров активных части- металлов и их оксидов.
Лекция д.х.н. О.П. Криворучко (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) была посвящена пост - синтетическому модифи-ированию HZSM-5 двухзарядными катионами, лекция д.х.н. Ё.М. Мороз (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) - применению модельных кривых радиального распределения электронной плотности как метода полного охарактеризования фазового состава и изучения локальной структуры.
Кинетическим методам исследования гетерогенных каталитических реак-ий и моделированию каталитических процессов были посвящены лекции к.х.н. Е.М. Садовской и к.х.н. В.И. Елохина (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск). К.х.н. С.И. Решетников (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) прочитал лекцию об автоматиза-ии химико-технологических расчетов.
Использование катализа для процессов тонкого органического синтеза было отражено в лек-ии к.х.н. О.А. Холдеевой (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск), а развитие уникальных проблем синтеза органических соединений на допланетных этапах эволюции - в лекции к.ф-м.н. В.Н. Снытникова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск).
В лекции к.х.н. О.В. Климова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) было рассказано о проблемах и приоритетах российского нефтеперерабатывающего комплекса, а также о разработках новых каталитических технологий получения высокосортных моторных топлив. Лекция д.х.н. Т.В. Андрушкевич (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) была посвящена вопросам дизайна гетерогенных катализаторов селективного окисления органических соединений в карбоновые кислоты, д.х.н. Н.М. Добрынкина (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) - дизайну гетерогенных катализаторов для про-ессов очистки промышленных сточных вод, д.х.н. В.А. Садыкова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) - новым перспективным процкссам получения синтез - газа при малых временах контакта.
Много вопросов задавали молодые ученые лектору к.х.н. В.Л. Кузнецову (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск). Тема его выступления - синтез новых катализаторов с заданными свойствами, которые позволяют получать в определенных условиях наноструктуры на основе различных материалов, начиная с углерода и кончая нитратом бора, карбидом титана, карбидом кремния и т.д. В данном случае молекулярный дизайн катализаторов играет особо важную роль - сама по себе каталитическая части-а очень мала и образуемая структура - тоже. Но именно размер главным образом и влияет на свойства катализаторов: изменяя их в незначительной степени, можно получать конечные продукты разного назначения.
Биокатализаторам и реакторам для гетерогенных биотехнологических процессов, дизайну биокатализаторов методами молекулярной эволю-ии были посвящены лек-ии к.х.н. Г.А. Коваленко (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) и д.б.н. С.Н. Загребельного (Новосибирский государственный университет).
С большим вниманием были выслушаны лекции, посвященные базам данных для специалистов в области катализа (к.х.н. Н.В. Круковская, Институт органической химии им. Н. Д. Зелинского РАН, Москва) и стратегии использования интеллектуальной собственности (М.Б. Демидов, Институт катализа им. Г.К. Борескова СО РАН, Новосибирск).
Представленные молодыми учеными устные и стендовые доклады оказались очень содержательными и интересными. Преподаватели задавали им много вопросов, вносили коррективы, поправляли и направляли, подводя к правильным выводам. По окончании конферен-ии были проведены УКруглые столыФ по деятельности Советов молодых ученых, по проблемам защиты интеллектуальной собственности.
На заключительном заседании конференции были подведены итоги объявленных конкурсов и одобрена идея продолжения проведения подобных Школ-конферен-ий молодых ученых раз в два года. Организаторы и участники выразили признательность Российскому фонду фундаментальных исследований за поддержку, которая позволила молодым сотрудникам принять участие в конферен-ии.
Т.В. Замулина
В период с 28 августа по 1 сентября 2005 года в столице Болгарии - Софии проходила работа 7 Европейского конгресса по катализу- EUROPACAT-VII. Этот международный форум, проходящий раз в два года, организуется Международной Федерацией Каталитических обществ. На этот раз организаторами Конгресса выступали балканские страны - Болгария и Греция, представленные своими каталитическими сообществами, а работу Оргкомитета возглавлял ученый с мировым именем - профессор Л. Петров, директор Института катализа Болгарской Академии наук.
В работе Конгресса приняло участие около 1000 человек, что достаточно близко к среднему числу участников за последние годы. Заседания происходили в Конгресс-центре, расположенном в Национальном дворце культуры в центре Софии.
Работа 7 Конгресса по катализу была построена по традиционной схеме - пленарные заседания, на которых заслушивались лекции приглашенных Оргкомитетом специалистов, секционные ключевые и устные доклады и стендовые сообщения. Относительно новая форма - флеш-презентации. Они предполагают краткое устное сообщение (1-2 слайда) и обсуждение у стенда. Сама по себе идея неплоха. Однако, практика еще не отработана, докладчики стремятся сказать как можно больше за очень ограниченное время (а надо бы как раз наоборот!). Возникает суета, а ведущие и аудитория больше сосредоточены на соблюдении регламента, чем на содержании презентаций.
Работа шла в четырех параллельных секциях. Это давно сложившаяся практика, позволяющая уложить огромную программу Конгресса в разумное время. При этом, однако, очевидна (но неизбежна) невозможность посетить доклады, читаемые одновременно в разных аудиториях. Участникам Конгресса с таким положением вещей приходилось мириться и оптимизировать посещение докладов в зависимости от научных интересов.
Организаторы и программный комитет EUROPACAT-VII пошли по пути формирования секций (симпозиумов) не по типу каталитических процессов, а по разделам науки о катализе и прикладным аспектам:

Такое построение программы Конгресса, конечно, имеет свои преимущества. Однако, оно же создает и определенные сложности для участников, интересующихся процессами определенного типа, например, окислением. Доклады (устные и стендовые) по данной тематике были представлены почти во всех секциях, иногда одновременно. По этой причине и вообще ввиду обширности программы Конгресса любой, а тем более краткий обзор неизбежно носит печать профессиональных интересов авторов этого материала.

Приятным сюрпризом для российской делегации оказалось то, что первым пленарным докладом, открывающим научные сессии Конгресса, стал доклад россиянина В.Н. Пармона - директора Института катализа СО РАН (Новосибирск) "Катализ в возобновляемой и нетрадиционной энергетике". Этот доклад касался существующих современных тенденций по применению каталитических процессов для (1) получения тепла из низкокалорийного или нетрадиционного горючего сырья; (2) преобразования возобновляемого растительного сырья (биомассы) в высококачественные топлива; (3) использования ядерной и солнечной энергии; (4) повышения эффективности преобразования химической энергии топлив в электрическую или механическую энергию, и (5) аккумулирования и использования разнообразных доступных источников бросовой низкопотенциальной теплоты, включая суточные и сезонные перепады температур. Большинство конкретных примеров по использованию обсуждаемых каталитических подходов к новому направлению энергетики, которым, по-видимому, суждено большое будущее, было продемонстрировано результатами экспериментальных исследований в Институте катализа СО РАН. При этом было обращено внимание на то, что если в момент инициирования этих работ лет 20-30 назад на проблему использования катализа для нетрадиционной энергетики смотрели, в основном, как на игрушку, то сейчас к этой проблеме стали относиться очень серьезно. Действительно, почти все ключевые доклады, прозвучавшие на Конгрессе в первый день, подробным образом раскрывали проблемы, поставленные в обсуждаемом пленарном докладе. Более того, почти 20 % всех сообщений, представленных на Конгрессе, так или иначе касались именно темы применения катализа в возобновляемой и нетрадиционной энергетике.
Можно отметить, что в значительной степени актуальность того или иного доклада отражала заполненность аудитории. Совершенно очевидно, что полные залы собирали пленарные лекции (В.Н. Пармон, М. Ньюрок, Р. Ламберт, Б.С. Клаузен, Р. Принс). Но также, например, полный зал собрала ключевая лекция Г.Р. Меима (Dow Benelux N.V./Dow Chemical Co.), посвященная в значительной мере состоянию проблемы перевода (получения) ценных полупродуктов органического синтеза (метанол, низшие олефины) из природного газа (метана, этана). Рассмотрены "движущие силы" и возможные направления такого перевода, ближайшие и перспективные цели, имеющиеся достижения и остающиеся нерешенные проблемы.
Вообще вопросам переработки легких парафинов было посвящено значительное число докладов, представленных на "нефтехимической" секции. Вполне очевидно, что это стимулировано ростом цен на нефть, что побуждает промышленность искать альтернативные источники углеводородного сырья, такие как природный газ, попутный газ и даже биогаз. Так, в ключевой лекции А. Хоека (Шелл Глобал Интернейшнл, Голландия), рассмотрены технологии переработки природного газа в жидкое топливо, разработанные данной компанией. Они включают получение синтез-газа и реакцию Фишера-Тропша с последующим гидрокрекингом/гидроизомеризацией полученных углеводородов. Получаемый продукт, в основном, предназначен для транспортного сектора, так что ключевая цель - обеспечение энергетической безопасности стран, где такая технология будет внедряться.
Отрадным можно считать включение в его программу работы специальной секции по кинетике каталитических процессов. К сожалению, тот факт, что катализ - суть в первую очередь кинетическое явление, как-то постепенно отошел на задний план с развитием сложных инструментальных методов исследования катализаторов. Т.е. наука о каталитическом процессе все более вытесняется наукой о каталитически-активных (или перспективных) материалах. Даже в работах, называющихся "Механизм реакции ..." речь зачастую идет о превращениях в катализаторе в большей степени, чем о реакции как таковой. Возможно, такой поворот лицом к кинетике в какой-то степени - отражение профессиональных интересов председателя Оргкомитета Конгресса проф. Л. Петрова, который представил вниманию аудитории (также переполненной) обзорную лекцию о месте кинетических исследований в изучении каталитических процессов.
Вспоминается совет-замечание проф. Дж.М. Томаса, сделанное за два года до этого на предыдущем Европейском Конгрессе по катализу: увлекаясь сложными методами исследования, не стоит забывать о простых вещах. При этом имелись в виду именно кинетические методы исследования. Это нашло яркое подтверждение в докладах на EUROPACAT-VII: нестационарные и изотопные методы, моделирование вновь демонстрируют свою силу при исследовании тонких деталей механизма каталитических процессов.
Отрадно и то, что именно на этой секции очень достойно были представлены доклады российских участников (М.М. Слинько, В.Ю. Бычкова, В.А. Садыкова, Э. Сульман и др.).
Кинетические методы были широко представлены и в докладах на секции, посвященной механизмам каталитических реакций. В наиболее интересных докладах здесь продемонстрировано, что "кинетический подход" отнюдь не находится в противоречии с "инструментальным". Ярким примером такого рода является представленное Р. Бёрчем элегантное исследование, в котором использовано сочетание спектральных (DRIFTS) и кинетических (SSITKA) методов. Показано, что так называемые "in situ" спектральные исследования реакционной способности поверхностных соединений в отсутствие каталитической реакции могут приводить к ошибочным выводам о том, через какие промежуточные соединения на самом деле протекает та или иная реакция. В частности, для прямой и обратной реакций паровой конверсии СО показано, что если при исследовании адсорбированных на поверхности групп реакционная способность, например, формиатов, намного выше, чем карбонатов, то в условиях стационарной каталитической реакции те же формиатные группы являются тупиковыми соединениями, превращаясь намного медленнее, чем карбонатные и карбонильные комплексы.
В работе К. Коста (C. Costa) с соавторами рассмотрена реакция парового реформинга фенола на нанесенных родиевых и железных катализаторах. Изотопно-динамические эксперименты с использованием меченых кислорода и водорода позволили обнаружить быстрый перенос (spillover) кислорода и водорода с активного компонента на носитель и обратно.
Кислородный обмен на оксидах церия/циркония, промотированных платиной, изучался в работе В. Садыкова (V. Sadykov) с соавторами. Проведенное исследование позволило объяснить промотирующий эффект платины на кислородный обмен быстрым переносом (spillover) кислорода с носителя и обратно, а также оценить константы скоростей и энергию активации реакции.
Попытка изучения сложной каталитической системы, представляющей собой смешанные оксиды молибдена-ванадия-вольфрама, с использованием метода SSITKA предпринята в работе Г. Фогеля (H. Vogel) с соавторами при изучении реакции селективного окисление акролеина. В результате была сформулирована макрокинетическая модель процесса, описывающая как протекание реакции, так и процессы кислородного обмена с катализатором.
На Конгрессе было представлено несколько докладов по эпоксидированию олефинов. Жидкофазное эпоксидирование с использованием органических гидропероксидов рассмотрено в работе Д. Фиерро (J. Fierro) с соавторами. Отмечено, что скорость эпоксидирования, протекающего на титанполисилоксановом катализаторе, зависит от природы титанового предшественника. Интересная работа по газофазному эпоксидированию пропилена на нанесенном оксиде железа была представлена М. Хронеком (M. Hronec) с соавторами. Добавляя в реакционную смесь аммиак или закись азота, авторы смогли добиться 10 %-ного выхода пропиленоксида. Предполагается, что аммиак выступает в качестве со-восстановителя, удаляя второй атом кислорода после встраивания первого по двойной связи пропилена, тогда как в случае N2O не исключено эпоксидирование за счет кислорода закиси азота. Другим примером интереса к реакции получения пропиленоксида может служить работа Т. Виссера (T. Visser) с соавторами, в которой рассматривается роль золота в катализаторах эпоксидирования на основе оксида титана. Показано, что золото участвует в активации пропилена, который затем реагирует с кислородными частицами на поверхности TiO2. Также заслуживают внимания работы по изучению реакции эпоксидирования этилена на серебре. Например, в работе В. Бухтиярова (V. Bukhtiyarov) с соавторами для исследования эпоксидирования в условиях in situ впервые был применен метод РФЭС высокого давления в сочетании с масс-спектрометрическим анализом газовой фазы. В работе К. Матиса (K. Matis) рассмотрено влияние пористой структуры и кислотности носителя на активность нанесенных серебряных катализаторов.
Большое количество работ, представленных на Конгрессе, были посвящены производству водорода, причем на нескольких симпозиумах сразу. О значительном интересе к данной тематике говорит тот факт, что более половины работ в секции "Новые каталитические процессы и материалы. Катализ новых источников энергии" относились к переработке углеводородного сырья в синтез-газ, паровой конверсии СО и селективному окислению СО в водородсодержащих смесях - основных стадиях получения водорода. Например, в ключевой лекции, представленной X. E. Verykios (Production of hydrogen by reformation of bio-fuels) было рассказано о переработке био-топлив в водород. Основное внимание в лекции было уделено конверсии биоэтанола на Ni и Ru содержащих катализаторах, нанесенных на La2O3 и Al2O3. В нескольких устных докладах были рассмотрены проблемы получения синтез- газа из метана на Rh, Ni и Cu- содержащих катализаторах. В ряде работ для реакции паровой конверсии метанола в качестве наиболее активного катализатора был предложен медно-цериевый катализатор, промотированный различными оксидами Zr, Mg, Gd, Y, Ga и др. Также во многих работах было показано, что медно-цериевый катализатор является активным и в реакции селективного окисления СО в присутствии водорода. Для реакции паровой конверсии СО было показано, что одними из наиболее активных и перспективных катализаторов являются Pt-содержащие системы, нанесенные на оксиды алюминия, титана, и церий - циркониевый твердый раствор. Огромный интерес был проявлен к работам, посвященным разработке микроструктурированных реакторов. Например, в работе Реброва Е.В. с соавторами был рассмотрен микрореактор с Mo2C катализатором в реакции паровой конверсии СО.
Следует отметить, что применение микрореакторов представляет значительный интерес не только для реакций получения водорода, но и для других областей катализа. Так, на Симпозиуме 8 в ключевой лекции C. De Bellefon (Лион, Франция), рассмотрено применение мезо- и микроструктурированных реакторов и компонентов в тонком органическом синтезе. Приведенные примеры включали изомеризацию аллильных спиртов в карбонильные соединения, асимметричное гидрирование двойной С=С связи и т. д.
Среди исследований в области науки о поверхности можно отметить работы, где данные, полученных на монокристаллах в условиях высокого вакуума, используются для описания кинетики процессов при реальных условиях. Так, в работе М.М. Слинько (Институт химической физики, Москва, Россия) совместно с B.E. Nieuwenhuys (Leiden University, the Netherlands) представлены результаты изучения природы кинетических автоколебаний реакций восстановления N2O водородом и СО на грани Ir(110). Предложенные авторами математические модели практически количественно воспроизвели область существования автоколебаний и основные особенности экспериментально наблюдаемых автоколебательных процессов. Так, наблюдаемый в эксперименте фазовый сдвиг между максимумами образования продуктов реакции (CO2, H2O и N2) удалось объяснить в рамках предлагаемых моделей ускорением процесса разложения N2O в присутствии адсорбированных атомов кислорода.
Доклад В. Ю. Бычкова (Институт химической физики, Москва, Россия) был посвящен изучению механизма автоколебаний в реакции окисления метана на никелевых катализаторах. Показано, что максимум и минимум веса катализатора соответствуют окисленному и восстановленному состояниям катализатора, соответственно. Предложен механизм автоколебаний, согласно которому автоколебательное поведение системы обусловлено периодическим окислением и восстановлением поверхности никеля. Экспериментальные данные положены в основу расчетов, которые объяснили основные особенности протекания реакции в автоколебательном режиме.
J. Libuda (Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin, Germany) представил результаты изучения методами STM, PES, TPD, IRAS (infrared reflection adsorption spectroscopy) и молекулярных пучков активности различных форм кислорода на наночастицах палладия, нанесенных на оксидный носитель (пленки оксида железа). Обнаружены качественные отличия процессов взаимодействия кислорода с поверхностью монокристаллов и наночастицами. Например, наблюдали частичное окисление поверхности наночастиц, при котором поверхностный оксид выступает в качестве резервуара атомов кислорода.
Изучение активности палладия в разложении и окислении метанола при давлениях ~ 0,1 мбар провели В.В. Каичев, И.П. Просвирин и В.И. Бухтияров (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия). Применение методов in situ РФЭС и масс-спектрометрии позволило обнаружить два маршрута разложения метанола на грани Pd(111). Результаты работы свидетельствуют об отложении углерода при температурах ниже 400 К, что является причиной низкой активности палладиевых катализаторов в реакциях дегидрирования метанола.
Взаимодействие нанесенных наночастиц палладия с атомами водорода, кислорода и углерода изучали G. Rupprechter с коллегами (Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin, Germany). Исследования проводили методами SFG (sum-frequency generation spectroscopy), PM-IRAS (polarization-modulation infrared reflection adsorption spectroscopy), TDS и XPS как в условиях сверхвысокого вакуума, так и при атмосферных давлениях с применением газового хроматографа. Обнаружено, что образование гидрида происходит главным образом на дефектах и фасетках (100) наночастиц. Окисление Pd-частиц также значительно зависит от наличия дефектов на поверхности.
Лекция L. Guczi (Chemical Research Centre of the Hungarian Academy of Sciences, Budapest, Hungary) была посвящена описанию одного из мощных и все чаще применяемых методов in situ исследования поверхности - метода sum-frequency generation (SFG) спектроскопии. Приведены примеры применения метода для изучения: адсорбции алкилгидроксамовой алкилфосфоновой кислот на медь и железо, адсорбции протеинов на титановые поверхности, структуры и свойств моно- и биметаллических монокристаллов и наночастиц, взаимодействия ионного и электронного пучков с полиэтиленом, границы раздела газ/жидкость пиридина, свойств модифицированных полимерных пленок и стекол.
Достаточно много работ было также посвящено применению спектроскопии ЯМР и ИК для исследования катализаторов и каталитических процессов, причем эти доклады были представлены на самых различных секциях.
Так, в докладе О2-04, представленном А. Жедоном (A. Gedeon, Universite Pierre et Marie Curie, Paris) продемонстрирована возможность применения гиперполяризованного ксенона для 129Xe ЯМР анализа микропористой структуры катализаторов. Лазерная гиперполяризация ядер 129 Xe приводит к резкому повышению чувствительности метода 129Xe ЯМР, что позволяет проводить анализ пористости катализатора с малым количеством адсорбированного ксенона. В этом случае химический сдвиг ксенона определяется исключительно его взаимодействием со стенками пористого материала, что делает возможным производить количественное определение размеров микропор в цеолитах и мезопористых материалах на основании зависимости хим. сдвига от внешнего давления ксенона над исследуемым материалом.
В докладе О2-25 Н. Малики (N. Malicki, Universite de Caen, Caen Cedex) продемонстрирована эффективность комбинированного использования ИК и ЯМР спектроскопии для характеристики влияния катионов Na на кислотные свойства ультрастабильного цеолита Y (HNaUSY). Использование ИК и ЯМР позволяет контролировать кислотность цеолитного катализатора в зависимости от количества протонов, замещенных на катионы Na. При этом были также обнаружены новые кислотные ОН группы в содалитовой ячейке цеолита, не участвующие в обмене с катионами Na, но оказывающие заметное влияние на кислотность цеолита.
В докладе ОF3-15 А.Г. Степанова (Институт катализа СО РАН, Новосибирск) продемонстрирована возможность использования ЯМР спектроскопии высокого разрешения в твердом теле in situ при высокой температуре для установления механизмов каталитических реакций. Анализ кинетики водородного обмена методами 1Н и 13С ЯМР in situ при температуре 250 - 300 ° С для пропана, различным образом меченного изотопами водорода (2Н) или углерода (13С), позволил установить механизм водородного обмена для пропана на кислотной форме цеолита ZSM-5.
Особый интерес участников Конгресса вызвали доклады по использованию магнитной резонансной томографии для визуализации протекания реакции от слоя к слою катализатора внутри проточного реактора. Так, в ключевой лекции "Chemical mapping inside catalytic reactors", прочитанной ведущим специалистом в этой области, профессором Linn F. Gladden (UK), приводятся данные по визуализации распределения степени превращения метанола по слою катализатора в реакции его этерификации с уксусной кислотой, а также визуализированы особенности распределения продуктов реакции гидрирования ряда олефинов вдоль реактора и в его поперечных слоях. Примечательно, что ранее на Европакате не было ключевых лекций по данной тематике, что, несомненно, говорит о том, что данный метод начинает набирать все большую и большую популярность среди каталитиков. В своем докладе профессор Gladden представила и результаты работ, выполняемых в Международном томографическом центре СО РАН под руководством д.х.н. И.В. Коптюга, что связано с тем, что данные работы внесли неоценимый вклад в развитие приложений метода ЯМР томографии в катализе. В группе профессора Gladden продолжают начатые в МТЦ СО РАН работы по исследованию гетерогенных каталитических реакций методом ЯМР томографии in situ. Тот факт, что метод ЯМР томографии позволяет не только визуализировать распределение жидкой фазы внутри пористого объекта, но и идентифицировать различные химические соединения на основе различия в их ЯМР спектрах, позволил исследовать гетерогенные каталитические процессы изомеризации и гидрирования октена-1 в слое катализатора, детектируя пространственно разрешенные 13С ЯМР спектры, используя естественное изотопное содержание данного изотопа. Ими также продолжаются работы по исследованию методом ЯМР томографии автокаталитических реакций, в частности, реакции Белоусова-Жаботинского, а также работы в области химической технологии - по исследованию потоков жидкостей в различных геометриях, структуры каталитических слоев в различных реакторах, в том числе в реакторе с псевдоожиженным слоем. Таким образом, в своей лекции профессор Gladden показала перспективность метода ЯМР томографии в области катализа и доказала необходимость развития данного метода применительно к каталитическим приложениям.
В докладе OF4-O3, представленном А.А. Лысовой (Международный томографический центр СО РАН и Институт катализа СО РАН) приведены данные по визуализации особенностей распределения жидкой фазы на катализаторе в проточном реакторе в ходе экзотермической реакции гидрирования α-метилстирола.
Работа катализаторов в реальных каталитических процессах всегда ставит задачи изучения стабильности работы катализатора, его дезактивации и регенерации. Рассмотрению этих вопросов был посвящен симпозиум №7.
В ключевой лекции (KN7-01)Дэвида Джексона (Англия) процессы, ведущие к дезактивации катализаторов, разделены на 3 типа - спекание, отравление и закоксовывание. Кроме того, отмечается важность механической стабильности катализатора, которая зависит от его механической прочности.
В зависимости от основной причины дезактивации катализатора, в докладах симпозиума предлагаются приемы его регенерации.
Так, для закоксовывающихся катализаторов (например, в процессах нефтепереработки) предлагается широко известный прием выжигания отложений (доклад KN7-01). Новым интересным приемом снижения закоксовывания катализатора во время его работы может быть проведение процесса под действием микроволнового излучения (07-06).
Для катализаторов, химически взаимодействующих с компонентами газовой смеси (вода, примеси), необходимо подобрать условия разложения образующихся соединений.
Обычно труднее всего регенерировать катализаторы со спекшимся активным компонентом. Среди интересных примеров регенерации спекшегося катализатора с восстановлением его дисперсности можно привести работу H. Birgsson (P7-02), в которой с целью увеличения дисперсности спекшегося платиноидного катализатора используется обработка его в газовой среде, содержащей кислород и хлор. Такая обработка приводит к образованию металл-хлоридных комплексов, приводящих к редиспергированию металла.
В дополнение к известным причинам дезактивации катализаторов, в работе Исуповой Л. (OF7-03) в качестве причины изменения селективности целевого процесса промышленного катализатора окисления аммиака ИК-42-1 рассматривается изменение его дефектной структуры, в частности - образование вакансий в структуре гематита, стабилизированного оксидом алюминия, и поверхностное обогащение родием. По-видимому, отжиг отработанного катализатора на воздухе может восстановить селективность катализатора. Кроме того, эта работа ставит задачу поиска допирующих примесей, стабилизирующих структуру гематита.
К уменьшению дезактивации катализатора может приводить и изменение условий протекания процесса. Так, замена неподвижного слоя катализатора на кипящий, а таблетированного на структурированный (блочный), использование переключающихся газовых потоков также могут способствовать увеличению срока работы катализатора (см. работы K. Al-Dalama OF7-06, N. Ghaloum Р7-13). Часто дезактивированный катализатор одного процесса может быть эффективен в других каталитических процессах (Р7-10).
Таким образом, глубокое изучение характера и причин дезактивации катализаторов необходимо не только для более правильного способа организации каталитического процесса, выбора правильного способа регенерации или возможности использования отработанного катализатора в других процессах, но также может послужить основой для разработки модифицированных катализаторов, увеличивающих стабильность к действию основного дезактивирующего фактора.
Симпозиум по экологическому катализу рассматривал такие достаточно традиционные проблемы, как удаление оксидов азота и серы из отходящих газов промышленных предприятий и автотранспорта, в том числе с использованием каталитического восстановления оксидов азота аммиаком и углеводородами (в том числе в присутствии водорода), а также катализаторов-адсорбентов; каталитическая очистка отходящих газов дизельных двигателей от сажи; катализаторы окисления летучих органических соединений и сжигания топлив; катализаторы для сжигания/ разложения/ гидродехлорирования хлоросодержащих углеводородов; удаление нитратов из воды. В области селективного каталитического восстановления оксидов азота углеводородами в избытке кислорода существенный прогресс в повышении низкотемпературной активности ряда катализаторов (например, серебро на оксиде алюминия) был достигнут при добавлении относительно небольшого количества водорода к реакционной смеси.
В ключевой лекции Г. Хатчинса на данном Симпозиуме под названием "Золотое будущее "зеленой" химии" рассмотрено применение нанесенных золотых катализаторов в таких процессах, как селективное окисление СО в избытке водорода, и синтез перекиси водорода из водорода и кислорода в атмосфере диоксида углерода при 2 оС в автоклаве, заполненном метанолом.
А. Генцлер (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) был представлен стендовый доклад, посвященный изучению жидкофазного гидродехлорирования хлорбензола и гексахлорбензола на нанесенных биметаллических катализаторах. Представленные результаты обсуждались и сравнивались с результатами работы S.A. Barta с соавторами (Португалия, Украина), которые представляли стендовый доклад по жидкофазному гидродехлорированию хлорбензола на палладиевых катализаторах на модифицированном углеродном носителе; J. Goralski с соавторами, которые представляли стендовый доклад по исследованию нанесенных палладиевых катализаторов в гидродехлорировании четыреххлористого углерода. На этом симпозиуме также был представлен ряд стендовых докладов, посвященных вопросам гидродехлорирования различных субстратов как в газовой, так и в жидкой фазах, и адсорбции хлорорганических соединений на модифицированных углеродных сорбентах из органических растворителей и воды.
На симпозиумах по приготовлению и охарактеризованию катализаторов был представлен широкий спектр устных и стендовых докладов по вопросам приготовления и свойств нанесенных палладийсодержащих моно- и биметаллических катализаторов.

Анализ представленных на конгрессе работ позволяет заключить, что катализаторы, получаемые в лаборатории исследования гидридных соединений Института катализа, по своим характеристикам не уступают мировым аналогам. Разработанные методики нанесения и восстановления предшественников активного компонента позволяют получать моно- и биметаллические катализаторы на основе палладия, обладающие высокой активностью и стабильностью в процессах газофазного и жидкофазного гидродехлорирования. Получаемые катализаторы позволяют проводить реакции в мягких условиях, без применения высоких температур и давлений, со стопроцентной конверсией субстратов.
На Симпозиумах 10-11 рассматривались проблемы дизайна новых каталитических материалов для топливных элементов разного типа, включая дизайн катодов и анодов. Среди этих докладов следует отметить ключевую лекцию Дж. Ирвина (Великобритания), посвященную дизайну каталитически активных анодов для твердоэлектролитных топливных элементов, работающих при пониженных (700-800 ° С) температурах. Показано, что сложные оксиды со структурой перовскита могут быть использованы как эффективные материалы для прямого окисления углеводородного топлива, включая пропан-бутан и более тяжелые углеводороды.
Спецификой данного конгресса, можно считать достаточно большое число работ, посвященных электрохимическому промотированию каталитических реакций, впервые продемонстрированному в работах Вайенаса (Греция), в основном представленных на Симпозиуме 11 по электрокатализу. По всей видимости, учет данного эффекта важен при дизайне новых материалов для топливных элементов.
Вцелом можно отметить, что 7 Европейский конгресс прошел успешно, и многочисленные обсуждения и дискуссии способствовали как развитию новых идей и подходов, так и критической апробации представленных результатов. Несомненно, что Конгресс также выполнил свою основную функцию - обеспечил контакты специалистов из разных областей катализа из разных стран не только Европы, но и Азии (Япония, Китай), и Америки.
Следующий 8 Европейский Конгресс по катализу состоится в Финляндии (Турку) в 2007 году.
М. Синев (Ин-т химической физики РАН, Москва)
В. Садыков, П. Снытников, А. Матвеев, А. Генцлер,
А. Степанов, Л. Исупова, А. Лысова, А. Сукнев,
В. Данилевич (Институт катализа, Новосибирск).
Фото Синева М.Ю., Пармона В.Н.
Gabor A. Somorjai Award for Creative Research in Catalysis
Sponsored by the Gabor A. & Judith K. Somorjai Endowment Fund
Ask D. Wayne Goodman about his interests and you have as much chance hearing about aviation as surface chemistry "Ever since I can remember, I was interested in flying," says Goodman, who holds the Robert A. Welch Chair in chemistry at Texas A&M University.
In his grade-school days, Goodman befriended a crop duster, who gave the young boy flying lessons in exchange for helping with chores at the airport in Greenville, Miss. The friend also worked as a stunt pilot on weekends, and as Goodman recalls, "on Saturday mornings, he'd pick me up and we would fly aerobatics together."
Asked how he managed to convince his parents to let him tag along for the ride while the pilot flew stunt maneuvers, Goodman answers, "I don't believe they knew." But they probably would have been supportive because enthusiasm for flying runs through the family. For example, Goodman's father Ö now 83 years old Ö continues to work as a flying instructor and aerial photographer.
At age 59, the younger Goodman also continues to fly. But most of his time during the past four decades has been devoted to science. In 1968, Goodman graduated from Mississippi College in Clinton with a bachelorÒs degree in chemistry. Six years later, he earned a Ph.D. degree in physical chemistry from the University of Texas, Austin.
GoodmanÒs keen interest in surface science was cultivated at the National Bureau of Standards (now the National Institute of Standards & Technology, NIST), where he served first as a postdoctoral associate and later as a staff scientist. Goodman notes that, while at NIST, he had the good fortune to work under the direction of two leading figures in surface science, both of whom are now professors of chemistry and physics. Some 25 years have passed since Goodman worked at NIST with John T. Yites Jr., now at the University of Pittsburgh, and Theodore E. Madey of Rutgers University, Piscataway, N.J., but Goodman continues to express great admiration and fondness for his mentors.
In 1980, Goodman accepted a position as a research scientist at Sandia National Laboratories, and from 1985 to 1988 he served as head of SandiaÒs Surface Science Division. In 1988, he moved to Texas when he was appointed professor of chemistry at Texas A&M.
With more than 400 scientific publications to his credit, GoodmanÒs contributions to surface science cover a broad range of topics. Early in his career, he developed techniques that made it possible to combine measurements of high-pressure reaction kinetics and high-vacuum surface analysis. The studies, which are often cited as textbook examples, led to a fundamental understanding of the roles of carbidic and graphitic forms of carbon in surface catalysis.
Goodman has also elucidated basic mechanisms through which small quantities of impurities can serve as catalyst poisons or promoters. He has also been a leader in the study of bimetallic catalysts and in developing procedures for applying high-resolution electron energy-loss spectroscopy and infrared reflection-absorption spectroscopy.
Experts worldwide regard Goodman as a scientific trendsetter. As a recent example, Charles T. Campbell, a chemistry professor at the University of Washington, Seattle, points to GoodmanÒs work on the size-dependent catalytic behavior of 2- to 3-nm gold particles. Campbell asserts that GoodmanÒs studies "caught the attention of the surface-science community and led to an explosion" of research in that subject. The award address will be presented before the Division of Colloid & Surface Chemistry.
HTTP://WWW.CEN-ONLINE.ORG
N & EN / JENUARY 31, 2005
ACS Award in Organometallic Chemistry
Sponsored by Dow Chemical Co.
Jack R. Norton, 59, professor of chemistry at Columbia University is best known for his work on the properties and reactions of transition-metal hydrides. It is work that has had a far-reaching impact on many of the most important processes in organometallic chemistry.
NortonÒs work on the kinetic and thermodynamic acidity of metal hydrides helped exploit the reactivity of metal hydrides as proton donors. He also carried out definitive studies of the kinetics of hydrogen atom transfer reactions of many of these same metal hydrides.
He has found examples of H+, H., and H- transfer processes and has studied how the rates of all three depend upon acidity (pKa), bond strength, and other thermodynamic parameters. He developed the first general procedure for measuring the pKa values of transition-metal hydrides and compared these thermodynamic acidities with the rates of proton transfer reaction of these hydrides.
He learned that metal acids behave much like carbon acids, with their proton transfer reactions proceeding slowly, owing to the requirement of substantial steric and electronic rearrangement.
Norton was among the first to demonstrate that the elimination of alkanes from some alkyl hydride complexes proceeds through a transient intermediate in which the alkane is coordinated to the transition metal. Such C-H complexes are pertinent to activation and functionalization of alkane C-H bonds by transition metals.
He provided early examples of cooperativity between metal centers in a cluster. Norton has used bimetallic systems to bridge the gap between homogenous and heterogeneous catalysts. He prepared a 1,2-dimetallacyclobutane and confirmed the prediction that ethylene elimination could not occur symmetrically
One of his colleagues notes, "A hallmark of NortonÒs research is that he does not shy away from challenging problems. ... He is assiduous in seeking comprehensive explanations for how reactions work."
A Dallas native, Norton received a B.A. in chemistry from Harvard University in 1967 and a Ph.D. in chemistry from Stanford University in 1972. His first academic position was at Princeton University where he was an assistant professor of chemistry from 1973 to 1979. He was an associate professor of chemistry and then a professor of chemistry at Colorado State University between 1979 and 1997. In 1997, he joined Columbia's faculty.
Norton has received a number of honors. In 1976, he was named a Dreyfus Foundation Teacher-Scholar. Between 1977 and 1981, he was an Alfred P. Sloan Fellow; in 1989, he became a Guggenheim Fellow; and in 1999, he became a fellow of the American Association for the Advancement of Science. He also received an Innovation Recognition Award from Union Carbide in 1985 and a Humboldt Research Award for Senior U.S. Scientists in 1993. In 1997, he received a Japan Society for the Promotion of Science Fellowship.
Norton was a member of the editorial advisory board for Organometallics between 1990 and 1994. He was treasurer of ACSÒs Division of Inorganic Chemistry between 1988 and 1991. From 1992 to April 2003, he served as an associate editor of the Journal of the American Chemical Society.
The award address will be presented before the Division of Inorganic Chemistry.
HTTP://WWW.CEN-ONLINE.ORG
Ñ & EN / JENUARY 31, 2005
IUPAC Prize for Young Chemists
The IUPAC Prize for Young Chemists has been established to encourage outstanding young research scientists at the beginning of their careers. The prize will be given for the most outstanding Ph.D. thesis in the general area of the chemical sciences, as described in a 1000-word essay.
AwardsIUPAC will award up to four prizes annually. Each prize will consist of USD 1000 cash and travel expenses to the next IUPAC Congress. In keeping with IUPACÒs status as a global organization, efforts will be made to assure fair geographic distribution of prizes.
Prizes will be presented biennially at the IUPAC Congress. Each awardee will be invited to present a poster on his/her research and to participate in a plenary award session.
Applications may be submitted, as described below, to the IUPAC Secretariat. In addition, some IUPAC National Adhering Organizations are soliciting applications in their own countries, frequently in conjunction with a national award. In such cases, application may be submitted to the NAO or to the Secretariat (not both). A list of NAOs is given below. Applications will be judged by a committee of eminent scientists appointed by the President of IUPAC.
Procedures for the 2006 Prize:
a. Entrants must have received their Ph.D. (or equivalent) degree, or completed all Ph.D. requirements including successful defense of the doctoral thesis, during calendar 2005 in any of the 65 countries that are Members or Associate Members of IUPAC. Entrants need not be citizens or residents of one of these countries at the time the application is submitted.
The research described in the entrantÒs thesis must be in the field of the chemical sciences, defined as "chemistry and those disciplines and technologies that make significant use of chemistry."
c. The IUPAC Prize recognizes only work that was performed while the entrant was a graduate student.
d. Application requires submission of a completed entry form, together with the material listed in items e and f. The entry form and supporting material should be submitted by e-mail whenever feasible. Additional material may be sent as needed by fax or mail.
e. An essay must be submitted by the entrant that describes his or her thesis work and places it in perspective relative to current research in the chemical sciences. The essay must be written in English by the entrant and may not exceed 1000 words. [For applications submitted through NAOs, a national language may be permissible, and the NAO will assist in translation to English. The announcement by the appropriate NAO should be consulted.]
f. Two supporting letters (sent by e-mail if feasible) are required from the thesis adviser and/or chairman of the thesis committee and one additional faculty member. These letters should comment on the qualifications and accomplishments of the applicant and the significance of the thesis work.
g. Complete applications must be received at the IUPAC Secretariat by February 1, 2006. If submitted through an IUPAC National Adhering Organization or Associate NAO, the deadline established by the NAO must be met. Early submission is strongly encouraged so any questions may be resolved before the deadline date.
IUPAC Secretariat
E-mail: secretariat@iupac.org
PO Box 13757
104 T. W. Alexander Drive, bldg 19
Research Triangle Park, NC 27709-3757, USA
Fax: +1 919 485 8706
URL: www.iupac.org
Participating Countries
Albania - Argentina - Australia - Austria - Bangladesh - Belgium - Brazil - Bulgaria - Canada - Chile - China/Beijing - China/Taipei -
Croatia - Cuba - Cyprus - Czech Republic - Denmark - Egypt - Estonia - Finland - France - Germany - Greece - Hong Kong - Hungary - India - Ireland - Israel - Italy - Japan - Kenya - Korea - Kuwait - Latvia -
Malaysia - Mauritius - Mexico - Netherlands - New Zealand - Norway - Pakistan - Peru - Philippines - Poland - Portugal - Puerto Rico - Romania - Russia - Saudi Arabia - Serbia and Montenegro - Singapore - Slovakia - Slovenia - South Africa - Spain - Sri Lanka - Sweden - Switzerland - Tanzania - Thailand - Tunisia - Turkey - Ukraine - United Kingdom - Uruguay - USA - Vietnam
Russia
Russian Academy of Sciences
Prof. Oleg M. Nefedov (Chairman),
National Committee of Russian Chemists,
c/o Vernadsky Institute of Geochemistry and Analytical Chemistry,
Kosygin Street 19, RU-119991 Moscow, Russia
Tel: +7 (095) 137 8625
Fax: +7 (095) 938 2054
E-mail: ncrc@mail.ru
www.ras.ru
Read more about it in Chem. Int. 25(1) 2003
XVII Международная конференция
по химическим реакторам ХИМРЕАКТОР-17
CHEMREACTOR-17
5-19 мая 2006 года Афины-Крит, Греция
Научная программа:
15-17 мая, Афины
18-19 мая, Крит
Пост-симпозиум КАТАЛИТИЧЕСКИЕ МЕТОДЫ
ИСПОЛЬЗОВАНИЯ ВОЗОБНОВЛЯЕМОГО СЫРЬЯ: ТОПЛИВО, ЭНЕРГИЯ, ХИМИЧЕСКИЕ ПРОДУКТЫ
Организаторы:
Срок подачи тезисов докладов - 20 января 2006 года.
Рабочий язык конференции - английский.
Детальная информация:
Оргкомитет конференции ХИМРЕАКТОР-17
Замулина Татьяна Владимировна
Институт катализа им. Г.К. Борескова СО РАН,
пр. Академика Лаврентьева, 5
630090 Новосибирск
Телефон/Факс: +7 (383) 330 62 97
E-mail: zam@catalysis.nsk.su
Announcement
Applied Physical Chemistry
3rd International Conference
Alushta, Ukraine, September 15 -20, 2006.
Dear Colleague
On behalf of the Scientific and Organizing Committee I am delighted to invite you to the 3rd International Conference on Applied Physical Chemistry, which will take place in Alushta, Ukraine, September 15 -20, 2006.
The Applied Physical Chemistry is recognized as an important international conference, which attracts scientists and engineers from the Former Soviet Union states and worldwide. Applied Physical Chemistry is started to develop in Ukraine since 2002. This field is intensively expanding being one of the priorities for the sustainable development of the National Economy. The Applied Physical Chemistry is the part of a series of successful conferences organized up to present.
The Applied Physical Chemistry conference is organized and co-sponsored by the Ministry of Education and Science of Ukraine, Academy of Sciences of Ukraine, V. Vernadsky Tavrida National University.
The conference should provide opportunities for exchange of the latest ideas and presentation and discussion of recent work on aspects of physico -chemical processes in ecosystems - water, air and soil and sustainable development including engineering and modeling. The aim of the conference is to encourage and facilitate the interdisciplinary communication among scientists, engineers, economists and professionals working in ecological systems and sustainable development.
The conference materials will be published in the Book of Abstracts and Conference Proceeding Book and will be available to conference attendees at the time of registration. In addition, the Proceedings will be widely distributed after the conference through the Internet and direct announcement to specialists and libraries and used in educational process. A poster session will be running, too.
We will also offer an attractive social program.
To register your interest as a participant at the conference or to submit an abstract for consideration, please complete and return the appropriate enquiry form (see enclosed Calls for Papers) to the Conference Secretariat, send your submission via e-mail or through the website. Full details are enclosed; please see the corresponding Call for Papers.
I hope that you will be able to attend the Applied Physical Chemistry International Conference and we are looking forward to welcoming you in Alushta, Ukraine, next 2006.
Yours sincerely,
Conference Secretariat
Conference Topics
A. Modern materials and technologies
Functional materials for fiber optics, optoelectronics and information storage
Nanomaterials
Sorption and membrane materials
Supramolecular structures
B. Physical Chemistry of biosphere processes
Mechanisms of pollutants mass transfer on the interface
Red/ox Processes in Natural Water, Atmosphere and Soil
Technologies of Natural Water, Atmosphere and Soil decontamination
Quality Standards, Control and Management of the environmental objects
Sustainable Development
C. Ecotoxicology
Impact of Chemicals on Human Health and Environment
Food Quality Control
Chemical Risk Analysis
Chemical Risk Mitigation
D. Applied Electrochemistry
Electrochemical methods of environmental protection, natural and wastewater treatment and conditioning
Ecological problems in electrochemical production
Cells and electrodes
E. Physico-Chemical methods for Environmental Monitoring
Monitoring of Water Quality and Environmental Modeling
Air Pollution Modeling, Quality Control and Management
Soil Contamination Control and Pollution Prevention
Persistent Organic Pollutants Management
Conference Secretariat Address:
V. Vernadsky Tavrida National University,
Ministry of Education and Science of Ukraine,
Simferopol', V. Vernadsky avenue, 4, 95007, Ukraine
Tel.: (0652) 23-03-47; (044) 424-32-12 Fax: (0652) 23-23-10
E-mail: ecochemistry@mail.ru, Kazdobin@ionc.kar.net,
physchemconf@yahoo.com Web: http://www.crimea.edu
Dr. Katherine Pershina

