
Распоряжением Правительства Российской Федерации от 25 октября
2017 года № 2345-р присуждены премии
Правительства
Российской Федерации 2017 года в области науки и техники и присвоено
почетное звание лауреата премии Правительства Российской Федерации в
области науки и техники:
Лунину Валерию Васильевичу, доктору химических наук, профессору, академику Российской академии наук, декану факультета федерального государственного бюджетного образовательного учреждения высшего образования “Московский государственный университет имени М.В. Ломоносова”, руководителю работы, Ткаченко Сергею Николаевичу, доктору химических наук, профессору того же учреждения; Боевской Елене Александровне, ведущему специалисту общества с ограниченной ответственностью "НИАП-КАТАЛИЗАТОР", Вейнбендеру Александру Яковлевичу, начальнику катализаторного производства, Голосману Евгению Зиновьевичу, доктору химических наук, профессору, главному научному сотруднику, Дульневу Алексею Викторовичу, кандидату технических наук, техническому директору, Ефремову Василию Николаевичу, кандидату технических наук, доценту, главному специалисту, – работникам того же общества; Кругловой Марии Александровне, заместителю генерального директора общества с ограниченной ответственностью "ДЕЛЬТАПЛАСТ"; Ткаченко Илье Сергеевичу, кандидату физико-математических наук, первому заместителю генерального директора общества с ограниченной ответственностью Научно-внедренческая фирма "Тимис"; Полушину Алексею Павловичу, директору по производству акционерного общества "Новомосковская акционерная компания "Азот", – за разработку, промышленное производство и масштабное внедрение высокоэффективных катализаторов двойного назначения для синтеза химических продуктов, получения технологических и очистки выбросных газов на предприятиях химической, нефтехимической, металлургической, машиностроительной, пищевой, атомной, оборонной, медицинской и других отраслей промышленности, а также в сфере жилищно-коммунального хозяйства.
Поздравляем коллег с высокой наградой!
проф. В.Б. Фенелонов
(Новосибирский государственный университет)
 21 ноября 2017 исполнилось
150 лет со дня рождения выдающегося химика-каталитика мирового уровня Владимира Николаевича Ипатьева,
одного из основателей науки и практики гетерогенного катализа, ученого с необычной судьбой. Среди российских ученых его имя стоит в
одном ряду с
21 ноября 2017 исполнилось
150 лет со дня рождения выдающегося химика-каталитика мирового уровня Владимира Николаевича Ипатьева,
одного из основателей науки и практики гетерогенного катализа, ученого с необычной судьбой. Среди российских ученых его имя стоит в
одном ряду с
Славное имя В.Н. Ипатьева начало возвращаться на Родину лишь в последние десятилетия1-9. Кратко рассмотрим основные этапы его драматической жизни.
Этап 1 – до начала Первой мировой войны. Владимир Николаевич Ипатьев родился в дворянской семье 2 (21) ноября 1867 года в Москве на Пресне. Его отец Николай Александрович – благополучный архитектор, мать Анна Дмитриевна – высокообразованная, начитанная женщина, знала несколько иностранных языков и воспитывала детей в духе передовых взглядов интеллигенции того времени.
Владимир Николаевич был старшим из трех детей, после гимназии учился в двухгодичном Александровском военном училище, затем в Михайловском артиллерийском училище и Михайловской артиллерийской академии. Выбор такого “артиллерийского” образования был связан с общим высоким уровнем преподавания именно в артиллерийских учебных заведениях того времени, где работали выдающиеся математики, физики, химики.
В Артиллерийской академии В.Н. Ипатьев настолько увлекся химией, что после ее окончания в 1892 году был оставлен там репетитором и помощником заведующего химической лабораторией. В том же году выполнял работы по органической химии в Санкт-Петербургском университете у проф. А.Е. Фаворского, а для повышения химического образования прослушал в университете курс лекций проф. Р.А. Меншуткина и проштудировал курс проф. А.М. Бутлерова. Первая научная работа Ипатьева – исследование изомеризации углеводородов – выполнена под руководством проф. А.Е. Фаворского. В 1894 году Ипатьев сделал свой первый доклад на заседании Русского физико-химического общества при Академии наук (РФХО), где бывший руководитель бутлеровской лаборатории проф. М.Д. Львов публично пожелал ему продолжать исследования с таким же успехом.
В 1895 году В.Н. Ипатьев защитил в Артиллерийской академии диссертацию “О действии брома на третичные спирты и бромистого водорода на ацетиленовые и алленовые углеводороды” и получил звание штатного преподавателя академии. За эту работу РФХО присудило Ипатьеву малую премию имени А.М. Бутлерова. В 1899 году – он экстраординарный, а с 1902 года – ординарный профессор химии, с 1909 года – заведующий химической лабораторией Артиллерийской академии.
С
1900 года В.Н. Ипатьев начал исследования в области гетерогенного катализа. После первого сообщения на январском заседании РФХО в 1901
году делает обстоятельный доклад о каталитическом разложении спиртов. В том же году его доклад “О двойном каталитическом разложении
алкоголей” стал центром внимания на семинаре Немецкого химического общества и Х съезде русских естествоиспытателей и врачей.
На завершающем банкете Х съезда ученик А.М. Бутлерова, старейшина российской каталитической школы
В последующие годы основным научным направлением работ Ипатьева стал катализ при высоких температурах и давлениях в среде водорода. В 1904 году была изобретена знаменитая “бомба Ипатьева” – надежный автоклав для исследований при высоких давлениях. Его важнейшие работы до 1917 года: термокаталитические реакции превращения спиртов, в которых предложены новые методы синтеза альдегидов, эфиров, олефинов, а позднее и диеновых углеводородов; исследования каталитических свойств оксида алюминия, причем именно он первым ввел в катализ оксид алюминия, ставший теперь основой многих распространенных катализаторов и носителей; циклизация и полимеризация олефинов (этилена, изобутилена и др.) – именно Ипатьев первым синтезировал полиэтилен; интермолекулярная гидрогенизация; синтез СН4 из СО и Н2; избирательное вытеснение металлов и их оксидов из растворов солей и др.
В 1909 году В.Н. Ипатьев первым установил принципиальную возможность получения бутадиена (дивинила) из этилового спирта на алюминиевом катализаторе, а в 1913 году первым осуществил синтез полиэтилена, первым начал применять многокомпонентные катализаторы, первым показал возможность совмещения окислительно-восстановительных и дегидратационных реакций в одном прямом процессе, использовал многофункциональные катализаторы при крекинге, риформинге и других процессах переработки нефти, разработал многочисленные промышленно важные процессы, такие как синтез полимербензинов на основе газообразных олефинов – отходов крекинга, алкилирование ароматических и парафиновых углеводородов олефинами для получения ценных химических продуктов и др.
Докторская диссертация, защищенная В.Н. Ипатьевым в Артиллерийской академии, формально не засчитывалась в гражданском академическом мире из-за
отсутствия соответствующего соглашения между академиями. Поэтому в 1908 году он успешно
защищает в Санкт-Петербургском университете теперь уже академическую докторскую диссертацию “Каталитические реакции при высоких
температурах и давлениях”. Диссертация получила высокую оценку проф. Д.П. Коновалова и проф. А.Е. Фаворского. За период с 1900 по
1917 годы Ипатьев публикует три монографии, два учебника и более ста статей в
российских и иностранных журналах. Одновременно совмещает свои исследования с преподаванием и работой в Артиллерийской Академии. В
1911 году ему присваивают звание генерал-майора, в 1916 году – генерал-лейтенанта, в 1914
году избирают членом-корреспондентом Академии наук, а в 1916 году – академиком. По приглашению
Наряду с фундаментальными исследованиями, В.Н. Ипатьев много внимания уделял химической технологии, практическому воплощению своих идей. В 1913 году его пригласили быть постоянным консультантом на Невском стеариновом заводе, где впервые в России была осуществлена гидрогенизация жиров. Затем поступили приглашения постоянно сотрудничать с нефтяной фирмой братьев Нобель, американской фирмой Дюпон и т.д. Он часто посещает химические заводы в России, а в загранкомандировках на различные международные научные форумы – и зарубежные заводы, куда его охотно приглашают для консультаций. Но 1 августа 1914 началась Первая мировая война.
Этап 2 – до ноября 1917 года. В начале войны оказалось, что производство боеприпасов в России не соответствует новым требованиям, в острейшем дефиците оказались взрывчатые вещества. Отечественные заводы не выпускали сырой бензол – важнейшее сырье для получения толуола, без которого нельзя получить многие взрывчатые вещества. В связи с этим в феврале 1915 года была организована Комиссия по заготовке взрывчатых веществ, возглавляемая В.Н. Ипатьевым. Первоочередные задачи комиссии: организация производства бензола и толуола пиролизом нефтепродуктов, получение серной и азотной кислот. Комиссия развернула энергичную деятельность, ее члены инспектировали заводы России, а Петрограде ежедневно собирались на квартире Ипатьева. Первый бензольный завод был пущен в августе 1915 г. – менее чем через полгода после утверждения планов его строительства, в конце года началось строительство около 20 новых небольших бензольных заводов, заводов по получению азотной и серной кислот и др.
Сначала Комиссия имела очень ограниченные полномочия и средства и должна была лишь проанализировать ситуацию в России и дать рекомендации. Но благодаря успешной работе Комиссия была преобразована в апреле 1916 года в Химический комитет при Главном артиллерийском управлении. Задачи комитета: организация производства порохов, взрывчатых веществ, фармацевтических препаратов, средств противохимической защиты и отравляющих веществ.
Этот Химический комитет и его руководитель были наделены уже широкими полномочиями с правом оперативного решения всех сложных вопросов непосредственно с начальником штаба Верховного главнокомандующего, военным и другими министрами, великими князьями – кураторами отдельных служб армии и промышленности, и периодически – с Николаем II, с которым Ипатьев неоднократно встречался не только на приемах, но и на завтраках, когда присутствовали всего несколько человек и решались самые больные вопросы.
Членами Химического комитета стали практически все виднейшие химики России: академик Н.С. Курнаков, профессора А.Е. Фаворский, А.Е. Чичибабин, Л.А. Чугаев, Г.В. Хлопин, В.Е. Тищенко и др., а В.Н. Ипатьев фактически стал главой химической промышленности России. Комитет осуществлял снабжение фронта продуктами военной химии (взрывчатые вещества, противогазы, отравляющие вещества, ГСМ, фармацевтика), имел контролеров-приемщиков на всех химических заводах, руководил строительством новых предприятий, в том числе первого в России завода по производству азотной кислоты окислением аммиака (пущен в 1917 г.). При поддержке Комитета Н.Д. Зелинский и Н.А. Шилов впервые в мире создали достаточно совершенный войсковой противогаз на древесном угле.
Заслуги ученого были отмечены как царем, так и правительственными и научными кругами. Ипатьев получил звание генерал-лейтенанта, а в 1915 году был избран в члены Российской императорской академии наук.
Этап 3 – с ноября 1917 года до смерти В.И. Ленина. В октябре 1917 года, внутренне не приняв Октябрьскую революцию и оставаясь по убеждениям сторонником конституционной монархии, В.Н. Ипатьев, тем не менее, встал на путь сотрудничества с советской властью. Решительно отказался от многочисленных предложений уехать на Запад или присоединиться к Белой армии. В 1921 году его посетил специально присланный эмиссар с предложением возглавить все белое движение, при этом ему гарантировали тайный выезд за границу и финансовую поддержку стран Запада. Ипатьев категорически отказался.
В ноябре 1917 года В.Н. Ипатьева навестил представитель Советского правительства Л.Я. Карпов, ведавший химической промышленностью. Выслушав обстоятельный рассказ Ипатьева об организации и деятельности Химического комитета, Карпов от имени правительства предложил ему сотрудничество в области демобилизации химической промышленности и ее дальнейшего развития. Ипатьев принял это предложение, сказав: “Я готов сделать все от меня зависящее, чтобы спасти созданную нами во время войны химическую промышленность”. В январе 1918 года он призвал членов Химического комитета к сотрудничеству с новым правительством. Ему пришлось приложить много усилий, чтобы убедить представителей русской науки отдать силы, знания и опыт строительству новой России и сохранить состав Химического комитета.
В декабре 1917 года В.Н. Ипатьев возглавляет Особую комиссию при Химическом отделе Высшего совета народного хозяйства (ВСНХ) по демобилизации и мобилизации химической промышленности (Л.Я. Карпов – член этой комиссии) и становится членом совета Главного управления артиллерии. В марте 1918 года, после беседы с Н.И. Подвойским, формировавшим в то время наркомат по военным делам, назначен постоянным членом и председателем Технического управления Военно-хозяйственного отдела Наркомата по военным делам, а в апреле – председателем комиссии по демобилизации и организации химической промышленности при правительстве.
В 1919 году Особая комиссия была преобразована в Технический совет химической промышленности при ВСНХ, а В.Н. Ипатьев был назначен председателем Совета. В эти же годы он активно участвует в создании новых институтов: в 1920 году выступил в качестве организатора и первого директора Государственного института научно-технических исследований (ГОНТИ) – это многопрофильный институт, координировавший важнейшие научно-технические работы в стране, организационное звено ВСНХ. Затем занимается организацией Института прикладной химии, Химико-фармацевтического института, Института удобрений и инсектофунгицидов, Радиевого института и др. В 1926 году ГОНТИ слился с Институтом прикладной химии, а в 1929 году из него выделился Институт высоких давлений, директором которого стал Ипатьев.
С мая 1921 года В.Н. Ипатьев становится членом Президиума ВСНХ и вскоре (после смерти Л.Я. Карпова) – начальником Главного химического управления ВСНХ (Главхима) и членом Госплана. Деятельность в качестве члена Советского правительства Ипатьев начал с докладной записки, содержащей подробный анализ состояния химической промышленности и перспектив ее развития. В качестве экстренной и крайней меры по восстановлению экономики страны и созданию мощной индустрии он предлагает привлечь иностранный капитал путем сдачи в концессию отдельных предприятий. А наряду с этим предлагает ввести так называемую реституцию – временное восстановление прав прежних владельцев, готовых сотрудничать с Советской властью. Интересно, что одновременно подобные предложения поступили и от старого большевика Л.Б. Красина, находившегося в то время на дипломатической работе. Бурное обсуждение записки Ипатьева на объединенном пленуме ВСНХ и Госплана завершилось полным одобрением.
По этому поводу и для обсуждения общих проблем развития химической промышленности В.Н. Ипатьев в 1921-22 годах несколько раз встречался с председателем правительства В.И. Лениным, который предложил ему лично выехать в заграничную командировку для переговоров о реституции и концессиях, сказав, что Советская власть вполне доверяет Ипатьеву и дает карт-бланш принимать кого угодно и вести переговоры, а в затруднительных ситуациях незамедлительно обращаться лично к Ленину и, если нужно, телеграфировать. В известной записке Н.П. Горбунову8 В.И. Ленин называет В.Н. Ипатьева “главой нашей химической промышленности“, а в 1922 году, когда из-за разногласий с тогдашним председателем ВСНХ П.А. Богдановым обсуждался вопрос о выводе В.Н. Ипатьева из членов Президиума ВСНХ и упразднении возглавляемого им Главхима, случайно узнавший об этом В.И. Ленин дал указание, чтобы “Ипатьев входил в состав Президиума ВСНХ при всяком числе его членов”.
В неоднократных заграничных командировках Ипатьев посещает ведущие заводы Германии, Франции, Италии, Англии, Бельгии и других стран, где ведет переговоры, заказывает научное и технологическое оборудование для химических заводов и институтов, активно участвует в работе научных форумов. И одновременно ежегодно публикует 20-30 научных работ.
В 1924 году Ипатьев был назначен председателем Главного химического управления при Революционном военном совете с важнейшей задачей – производство связанного азота. В том же году он выступает с предложением организовать Добровольное общество помощи развитию химии и химической промышленности (Доброхим). Инициатива была одобрена, Доброхим позже реорганизован в Авиахим, в 1927 – Осоавиахим СССР, затем – ДОСААФ. Первым председателем избран Л.Д. Троцкий, замами – В.Н. Ипатьев и М.В. Фрунзе. В задачи этой добровольной организации, наряду с задачами обороны, входили химизация сельского хозяйства, разъяснение важности химии в экономике, энергетике и т.д.
Этап 4 – с 1924 года до отъезда в эмиграцию. В мае 1927 года 60-летие В.Н. Ипатьева и 35-летие его научной деятельности были отмечены как крупное для всей страны событие, праздник мировой науки. Официальные торжества проводились в крупнейшем в те времена зале – в Политехническом музее в Москве, где в Президиуме находились члены правительства, ВСНХ, руководство РККА. Юбилей освещался в газетах, на радио, отмечался на собрания ученых и работников химической промышленности. Ипатьев получил звание заслуженного деятеля науки и премию имени Ленина.
В начале 1927 года Ипатьев получил предложение от руководителей Общества баварских азотных заводов провести у них совместные исследования по катализу при высоких давлениях. Изобретения, которые будут сделаны им в Германии, должны были патентоваться фирмой в Германии с указанием авторства Ипатьева, а в СССР он получал право патентовать их от своего имени, и по договору они безвозмездно переходили в собственность СССР. Советское правительство нашло предложения германской стороны приемлемыми и дало согласие на проведение Ипатьевым исследований в Германии с ежегодными отчетами на заседании Президиума ВСНХ. В июне 1929 года Президиум ВСНХ заслушал доклад Ипатьева о работе в Германии и признал, что она привела к чрезвычайно важным открытиям. Особо отмечалось, что созданная им в Ленинграде лаборатория высокого давления “становится уже в настоящее время школой химиков, работающих в области высоких давлений и температур, и в дальнейшем будет играть громадную роль в деле подготовки новых кадров работников в этой области”. В 1929 году эта лаборатория преобразована в Институт высоких давлений, который Ипатьев возглавлял до своего отъезда из СССР в 1930 году.
Несмотря
на успехи и триумфальный юбилей, в эти годы ситуация вокруг В.Н.
Ипатьева стала усложняться. В 1926 году его вывели из состава ВСНХ и
руководства химией по линии Красной Армии (Ипатьев узнал
об этом из газет).
Начались аресты коллег, близких друзей, учеников: арестованы
академики С.Ф. Платонов и
Этап 5 – эмиграция. В 1930 году В.Н. Ипатьев принимает твердое, но крайне тяжелое для себя решение – выехать за границу и до поры до времени не возвращаться. В июне 1930 года он получил персональное приглашение на II Международный энергетический конгресс в Берлине. Оформление его документов задерживалось, но одного из делегатов арестовали, место в делегации СССР освободилось (было всего 10 мест), и было неудобно оставлять его незаполненным при наличии персонального приглашения Ипатьеву. В июне 1930 года Владимир Николаевич вместе с женой Варварой Дмитриевной выехал в Берлин для участия в конгрессе, после которого получил разрешение правительства и АН СССР задержаться на лечение сроком на один год. В июне-августе 1930 года он побывал во Франции и Англии, в сентябре прибыл в США, сначала в Нью-Йорк, затем в Чикаго, где ему была сделана сложная операция на горле. Здесь же, в Чикагском университете, он стал читать курс лекций по катализу и одновременно приступил к экспериментальным работам по контракту с фирмой Universal Oil Products Co. в прекрасно оборудованной для него лаборатории.
Вплоть до 1936 года Ипатьев регулярно передавал в СССР результаты своих работ, выполненных в США, за свой счет закупал и отправлял научное оборудование для лаборатории высоких давлений, оплачивал зарубежные командировки сотрудников. В 1936 году в СССР и США одновременно вышла его фундаментальная монография “Каталитические реакции при высоких температурах и давлениях”, соответственно, на русском и английском.
В
1931 году отпуск Ипатьева был продлен на три года, но с 1935 года
Правительство СССР и Академия требуют его возвращения. А приходящие
из СССР сведения вызывают страшную тревогу. Усиливаются репрессии,
арестованы академики П.П. Лазарев, М.Н. Сперанский, подвергнут
шельмованию академик
В своих воспоминаниях Ипатьев пишет, что в 1927 году он был в гостях у нобелевского лауреата Вальтера Нернста в Германии, где встретился с Альбертом Эйнштейном: “Один из немецких профессоров спросил меня, почему я совсем не покину СССР и не переселюсь за границу для продолжения своих научных работ, где я найду, несомненно, гораздо больше удобств, чем у себя на Родине. Я в то время не имел ни малейшей идеи покинуть свою страну и ответил, что как патриот своей Родины, должен остаться в ней до конца моей жизни и посвятить ей все мои силы… Профессор Эйнштейн слышал мой ответ и громко заявил: “Вот этот ответ и я вполне разделяю, так и надо поступать”. И вот прошло 4-5 лет после этого разговора, и мы оба нарушили наш принцип: мы теперь эмигранты и не вернулись в свои страны по нашему персональному решению, а не потому, что были изгнаны нашими правительствами…”.
В 1938 году на свои сбережения Ипатьев основал Лабораторию катализа и высоких давлений при Нортуэстернском университете (город Эванстон, недалеко от Чикаго). Лаборатория существует до сих пор как Ipatieff High Pressure Laboratory at Northwestern University. Здесь он продолжал начатые в России исследования. В 1936 году В.Н. Ипатьев первым предложил каталитический крекинг, позволивший значительно увеличить выход бензина при переработке нефти. Вторым прославившим его изобретением стало получение высокооктанового бензина. И именно исследования Ипатьева по дегидрированию и полимеризации позволили наладить производство всевозможных полимеров и пластмасс, без которых современная жизнь невозможна.
Владимир Николаевич оставался директором своей лаборатории до последних дней жизни, одновременно преподавал в Чикагском университете, был консультантом в нефтяной фирме. Все заработанные деньги вкладывал в развитие лаборатории, приглашая на работу только знающих русский язык. В 1937 году Ипатьев был назван в США “Человеком года” (выбран из 1000 претендентов на это звание). В 1939 году его избрали членом Национальной Академии США, и в том же году в Париже состоялось торжественное вручение ему высшей награды Французского химического общества - Медали имени А. Лавуазье. В ноябре 1942 года, когда в США Американское химическое общество отмечало 75-летие со дня рождения и 50-летие научной деятельности В.Н. Ипатьева, нобелевский лауреат Рихард Вильштеттер заявил: “Никогда за всю историю химии в ней не появлялся более великий человек, чем Ипатьев”.
А как жил, чем дышал все эти годы “невозвращенец” Ипатьев, рассказал профессору В.И. Кузнецову, посетившему лабораторию Ипатьева в 1967 г., профессор Герман Пайнс, самый близкий из учеников, друг и душеприказчик В.Н. Ипатьева: “Вы, русские, совсем не представляете В.Н. Ипатьева, не понимаете даже, кем был Ипатьев. Каждый час своей жизни здесь, в США, каждый шаг в своей научной деятельности он отдавал России. Беспредельная любовь к Родине, какой я никогда и ни у кого из эмигрантов не видел, была той почвой, на которой произрастали все выдающиеся результаты его научной деятельности. Мы, его ученики, приобрели или построили для себя здесь, на берегу озера Мичиган, хорошие коттеджи. Владимир Николаевич не захотел иметь собственный дом. Он считал себя иностранцем и 22 года (!) жил, снимая номер в гостинице в Чикаго. Каждый из нас имел свою автомашину. Он же отказался от этого “акта оседлости” и пользовался казенным транспортом. Почти все обзавелись собственными яхтами, зачастую двухпалубными, для прогулок по озеру Мичиган. Владимир Николаевич отказался и от этого. Жил он довольно замкнуто, охотно общаясь только с близкими ему людьми; например, с удовольствием встречался с С.В. Рахманиновым. Он жил Россией, а русские платили ему нарочитым забвением. Я особенно возмущен поведением руководства Академии наук СССР. На 100-летие со дня рождения Ипатьева в июне 1967 года мы пригласили пять ученых из СССР, оплатили им дорогу в США и обратно и их пребывание здесь. Мне известно, что люди хотели приехать к нам, но их не пустили. Это вызвало негодование у гостей из Франции, Германии, Англии”.
Всю жизнь в США супруги Ипатьевы снимали скромный номер в гостинице, жили замкнуто. Их редкие письма родственникам в Ленинград проникнуты тоской по родным местам. Владимир Николаевич никогда не переставал интересоваться жизнью советских людей, достижениями советской науки. До глубины души его трогали поражения и победы Красной Армии в годы войны. Вместе с композитором С.В. Рахманиновым и другими известными эмигрантами был организован фонд Помощи Красной армии и народу СССР. А чтобы скрасить одиночество, Ипатьевы удочерили и воспитали двух русских девочек-сирот.
Начиная с 1944 года, Ипатьев трижды предпринимал попытки вернуться в СССР, но неизменно получал отказ от посла СССР в США А.А. Громыко. Последняя попытка была в 1951 году, ее реализации помешала тяжелая болезнь и смерть. До самого конца (29 ноября 1952 года), несмотря на преклонный возраст, В.Н. Ипатьев трудился в лаборатории. А на его могильной плите осталась надпись “In Memory of Russian Genius Vladimir Nikolaevich Ipatieff. The Inventor of Octane Gasoline”. Его супруга Варвара Дмитриевна пережила мужа всего на 10 дней и была похоронена рядом.
За 60 лет научной деятельности Владимир Николаевич Ипатьев написал около 400 статей, десятки книг, он автор более 200 изобретений, член Американской, Берлинской, Парижской, Геттингенской академий, лауреат премий В.И. Ленина, А.М. Бутлерова и др., медалей Уилларда Гиббса, Марселена Бертло, Антуана Лавуазье.
Наиболее
известные российские ученики В.Н. Ипатьева: академик Г.А.
Разуваев, чл.-корр. А.Д. Петров, профессора Б.Н. Долгов, А.В.
Фрост, В.В. Ипатьев, М.С. Немцев и др., а из иностранцев –
американцы
А
теперь попытаемся ответить на очевидный вопрос –
почему
такой выдающийся ученый как
Первопроходцем
разработки метода высокого давления (до 400-500 атм) по праву следует
считать
А в представлении В.Н. Ипатьева в члены Академии наук России (ноябрь 1915 года), которое составили академики П.И. Вальден, Б.Б. Голицин и Н.С. Курнаков, прямо указана чрезвычайная важность его исследований и отмечено, что они “отличаются большим разнообразием, чем работы П. Сабатье, удостоившегося в 1912 году Нобелевской премии”. Вот что писал более конкретно П.И. Вальден: “Если Сабатье получил Нобелевскую премию только за одну каталитическую реакцию, то работы Ипатьева несомненно заслуживают этой же премии, так как он гораздо шире применил катализаторы для различных реакций, ввел совершенно новый метод высоких давлений, что позволило вести гидрогенизацию с такими веществами, работать с которыми по методу Сабатье было невозможно”.
В опубликованной в 1997 году статье д.х.н. Ю.И. Соловьева, сотрудника архива РАН, высказано мнение, что причина того, что В.Н. Ипатьев не стал первым российским лауреатом Нобелевской премии по химии – в политических пристрастиях членов Нобелевского комитета, что “Нобелевский комитет по химии не пожелал присуждать премию ученому из-за его активного участия в экономическом развитии России в 1918-1927 годах”. По этой версии все решили политические пристрастия.
Но существует и более прозаическая версия, изложенная в фундаментальном историческом исследовании А.М. Блоха9: основная причина в том, что соотечественники, прекрасно осведомленные о выдающихся работах Ипатьева 1905-1913 гг., просто не удосужились представить его кандидатуру в Нобелевский комитет. Итак, соотечественники не вспомнили, а без письменных представлений кандидатуры в Нобелевском комитете не рассматриваются. А.М. Блох упомянул и собственную промашку В.Н. Ипатьева, который в докладе на Международном конгрессе по промышленной химии в Страсбурге (1928 г.) отметил, что “патенты Бергиуса (1911 г.) всецело основаны на моих работах, сделанных еще в 1903-1904 годах, и мой метод… был целиком применен для гидрогенизации смол и углей”. Но патенты все же получил Бергиус, а не Ипатьев, своевременно их не оформивший… Как бы там ни было, но отсутствие имени Ипатьева среди Нобелевских лауреатов – вопиющая несправедливость, которую, к сожалению, уже невозможно исправить.
Но в наших силах исправить другую несправедливость – вернуть славное имя нашему великому соотечественнику Владимиру Николаевичу Ипатьеву, которым мы должны гордиться. Он во все времена при любых обстоятельствах оставался патриотом России, и мы должны перед ним покаяться за вопиющие ошибки, которые совершили другие наши же соотечественники.
Литература
(к 150-летию академика В.Н. Ипатьева)
к.пед.н. С.В. Телешов
(Санкт-Петербург)
 В истории химии нашей страны
нельзя не вспомнить удивительного человека, который не имел
классического химического образования, т. е. университетского диплома по этой специальности. Тем не менее он
получил мировое признание. Это Владимир Николаевич Ипатьев. Осенью
2017 года отмечается 150 лет со дня рождения учёного, нашего
соотечественника.
В истории химии нашей страны
нельзя не вспомнить удивительного человека, который не имел
классического химического образования, т. е. университетского диплома по этой специальности. Тем не менее он
получил мировое признание. Это Владимир Николаевич Ипатьев. Осенью
2017 года отмечается 150 лет со дня рождения учёного, нашего
соотечественника.
Введение
Старший сын Владимир – первенец – в семье архитектора Николая Александровича Ипатьева родился в Москве на Средней Пресне. Как отметил в своих воспоминаниях сам В.Н. Ипатьев, “угольный дом Средней Пресни и Безымянного переулка” [1, c. 38]. Немного позднее семья переехала по соседству в собственный дом. В 1876 году он поступил в 5-ю классическую гимназию на Поварской улице. Непростые семейные обстоятельства, включая финансовые затруднения, привели к необходимости определить Володю со второго класса в 3-ю военную гимназию, в которой обучение получали за казённый счёт. Весной 1882 года Владимир приступил к изучению раздела о химических явлениях в курсе физики по учебнику К.Д. Краевича для 6-го класса военной гимназии [2, c. 589-610]. “Меня поразила, – пишет он в своих воспоминаниях, – стройная связанность описываемых явлений, – и я снова и снова перечитывал эти немногие страницы (раздел “Краткий очерк химических явлений” в этом учебнике занимал всего 21 страницу – Авт.), стараясь понять законы, которые эту связанность определяют. Конечно, я понял далеко не всё... Помню, больше всего меня заинтересовали законы постоянства состава и кратных отношений, а также атомистическая теория строения вещества. Мне казалось, что я впервые посмотрел на мир открытыми глазами, – и мне захотелось учиться, чтобы полнее и лучше его понимать... Теперь, когда я оглядываюсь на пройденный путь, мне ясно, что с этого момента интерес к химии стал основным для всей моей интеллектуальной жизни” [3, с. 20, 21, 30].
Врата учёности
После окончания гимназии, осенью1884 года, 16-ти с половиной лет отроду юноша навсегда покинул отчий дом и два года продолжал учёбу в 3-м Александровском московском пехотном училище. Здесь на младшем курсе был учебный предмет “химия”. Преподавали её курс по учебнику Потылицына (Потылицын Алексей Лаврентьевич (1845-1905), ученик Д.И. Менделеева, профессор. Его учебник «Начальный курс химии» издавался в 1881-1908 гг.). Преподавателем был Николай Павлович Нечаев (Нечаев Н.П. (1841-1917), генерал от артиллерии, автор учебников по химии. Один из учредителей РФХО. Автор объёмной периодической таблицы, построенной в виде тел вращения (усечённых конусов), называемой “Способ Нечаева”). Обучение было поставлено практически без лабораторных работ, и юнкер начал по своей инициативе дополнительно изучать химию по переводному учебнику Кольбе (Kolbe Adolph Wilhelm Hermann (1818-1884), химик, ученик Ф. Вёлера и Р. Бунзена, профессор). “Знакомство с книгой Кольбе помогло мне только в смысле лучшего понимания некоторых отдельных химических реакций” [3, с. 32].
 В
сентябре 1886 года молодой человек для продолжения обучения поступил
на 3-й курс Михайловского артиллерийского училища в Петербурге. К
сожалению, и здесь химия излагалась без разъяснений, требовалось лишь
зазубривание фактического материала. “Всё было как бы
сознательно направлено на то, чтобы убить в нас всякий интерес к
химии...” [3, с. 38]. Тем не менее, обладая великолепной
памятью и способностью к химии, Владимир
по этому предмету получил полный балл (в военных учебных заведениях
была принята двенадцатибалльная
система оценивания –
Авт.), был произведён в
офицеры и получил направление в артиллерийскую бригаду в подмосковный
Серпухов, где почти два года (1886-1888) длилась его строевая служба.
Ему, артиллерийскому офицеру, было настолько интересно заниматься
химией, что перед тем как прибыть на место службы, он потратил
существенную часть полученных при выпуске из училища денег на
приобретение маленькой домашней лаборатории качественного анализа
(потом в его жизни были и другие домашние лаборатории). Владимир стал
самостоятельно готовиться к поступлению в Михайловскую артиллерийскую
академию. Молодой поручик проделал колоссальную подготовительную
работу: осуществил самостоятельно почти все реакции качественного
анализа главнейших элементов; учебники не просто выучил, а продумал,
проверяя их выводы теми анализами, на которые эти выводы опирались;
вник в тайны Периодического закона и научился пользоваться
Периодической таблицей при изучении элементов.
В
сентябре 1886 года молодой человек для продолжения обучения поступил
на 3-й курс Михайловского артиллерийского училища в Петербурге. К
сожалению, и здесь химия излагалась без разъяснений, требовалось лишь
зазубривание фактического материала. “Всё было как бы
сознательно направлено на то, чтобы убить в нас всякий интерес к
химии...” [3, с. 38]. Тем не менее, обладая великолепной
памятью и способностью к химии, Владимир
по этому предмету получил полный балл (в военных учебных заведениях
была принята двенадцатибалльная
система оценивания –
Авт.), был произведён в
офицеры и получил направление в артиллерийскую бригаду в подмосковный
Серпухов, где почти два года (1886-1888) длилась его строевая служба.
Ему, артиллерийскому офицеру, было настолько интересно заниматься
химией, что перед тем как прибыть на место службы, он потратил
существенную часть полученных при выпуске из училища денег на
приобретение маленькой домашней лаборатории качественного анализа
(потом в его жизни были и другие домашние лаборатории). Владимир стал
самостоятельно готовиться к поступлению в Михайловскую артиллерийскую
академию. Молодой поручик проделал колоссальную подготовительную
работу: осуществил самостоятельно почти все реакции качественного
анализа главнейших элементов; учебники не просто выучил, а продумал,
проверяя их выводы теми анализами, на которые эти выводы опирались;
вник в тайны Периодического закона и научился пользоваться
Периодической таблицей при изучении элементов.
“Огромным счастьем было, что на русском языке существовали две такие замечательные книги, как “Основы химии” Менделеева и “Аналитическая химия” Меншуткина. Обе они имели огромное воспитательное значение, позволяя работать без преподавателя и приучая самостоятельно думать. Эти две книги и были моими действительными учителями химии. Я ежедневно прочитывал по несколько страниц из “Основ химии” и затем старался проделать в лаборатории те главные реакции, с которыми я только что познакомился. Параллельно я проделывал реакции... по качественному анализу Меншуткина... Этот метод изучения, который я нащупал тогда совсем интуитивно, без помощи с чьей-либо стороны... В настоящее время он является общепринятым” [3, с. 41] (увы, не в настоящее время – В.Н. Ипатьев пишет о периоде конца XIX – начала XX в. – Авт.).
В августе 1889 года исполнилась его заветная мечта – посвятить свои силы изучению химии – он стал слушателем Михайловской артиллерийской Академии. Ему шёл тогда двадцать второй год. В это же время Владимир Николаевич узнал о существовании Русского физико-химического общества и захотел стать его членом. Для вступления в РФХО следовало представить три рекомендации членов Общества. Преподаватели Академии Г.А. Забудский, М.А. Котиков, С.В. Панпушко поручились за него, и поручик был принят в ряды, объединявшие лучших химиков России (протокол от 29.11.1890) [4, с. 2].
Именно в эти годы состоялось его знакомство с Д.И. Менделеевым, Н.Н. Бекетовым и Д.П. Коноваловым. Интерес к химии, который Владимир Николаевич ни от кого не скрывал, его практический опыт, полученный в домашней и академической лабораториях, закономерно привёл его на старшем курсе к выполнению исследовательских работ под руководством инженера-технолога профессора Д.К. Чернова, читавшего в Академии курс металлургии (Чернов Дмитрий Константинович (1839-1921) – один из ведущих специалистов по сталеплавильному делу, профессор Михайловской артиллерийской Академии с 1889 г.). Завершилась эта работа докладами в Техническом обществе, в Русском физико-химическом обществе и публикацией его первой печатной научной работы в “Артиллерийском журнале” [5]. Ещё во время обучения он составил по просьбе сокурсников (!) рукописные записки по курсам качественного и количественного анализа (в библиотеке современной Михайловской Академии они не сохранились).
Окончив
курс Академии в мае 1892 года, В.Н. Ипатьев получил лестное
предложение остаться в Академии преподавателем и 12 августа
приступил к службе в качестве репетитора по химии в своей alma
mater.
Репетиторов-химиков называли “конно-химическим эскадроном”,
так как они числились по конной артиллерии. Ему предстояло воссоздать
химическую лабораторию в Академии (созданную в 1868 году
Сказано
– сделано. И двадцатипятилетний Владимир Николаевич посещает
лекции в университете по органической химии у Н.А. Меншуткина,
штудирует последний прижизненный курс лекций по органике (1884-1885
гг.), прочитанный А.М. Бутлеровым и его “Введение к полному
изучению органической химии”. Одновременно он работает в
маленькой лаборатории А.Е. Фаворского и приготовливает 4 кг
цинкметила,
В 1895 году успешно прошла защита диссертации “Действие брома на третичные спирты и присоединение бромистого водорода к алленам и двузамещённым ацетиленам”. В.Н. Ипатьев единогласно признан достойным звания штатного преподавателя академии (но не доктором химии). Два с половиной года непрерывной работы дали ему почувствовать, что “и моя капля мёда есть в сокровищнице химических знаний”! [3, с. 145]. Летом этого же года возникла мысль заняться изучением реакции присоединения бромистого водорода в уксуснокислом растворе к недавно выделенному из каучука и скипидара изопрену. А в начале 1896 года перспективный молодой учёный получает от Михайловской Академии право на 16-месячную стажировку за границей. Рекомендательное письмо к А. фон Байеру от Л.Н. Шишкова, который в бытность в Гейдельбергском университете стажировался у профессора Р. Бунзена вместе с А. Байером и был дружен с последним, помогло В.Н. Ипатьеву получить не только рабочий стол длиной два метра в лаборатории будущего Нобелевского лауреата, но и непосредственное руководство (его получали далеко не все стажёры) маститого химика. Он трудился столь плодотворно, что А. Байер предложил ему совместную публикацию по результатам работы, хотя в то время это было не принято [6] (Johann Friedrich Wilhelm Adolf von Baeyer (1835-1917), химик-органик. Нобелевская премия по химии вручена в 1905 г.).
Уже в следующем (1897) году во время этой стажировки в лаборатории А. фон Байера, наследовавшего лабораторию Ю. Либиха, В.Н. Ипатьев доказал наличие открытой цепи углеродных атомов в изопрене и первым в мире осуществил его синтез не из каучука [3, с. 180-181; 7; 8]. По окончании стажировки при фотографировании “на память” А. Байер лишь одного стажёра – В.Н. Ипатьева – пригласил занять место в первом ряду рядом с профессорами!

Сотрудники
и стажёры лаборатории Адольфа фон Байера, Мюнхен, 1897:
в первом ряду
(справа налево): В.Н. Ипатьев, проф. Кенигс,
проф. А. фон Байер,
проф. Тиле, д-р Вихорн, д-р Эйнхорн
Владимир Николаевич в это время получил от А. Байера такие ценные советы, как: 1) важно не знание огромного количества фактов, а основательное понимание основ науки и знание тех фактов, которые подтверждают наши теоретические воззрения; 2) ранее, чем делать реакции в большом масштабе, стараться попробовать их осуществить в пробирном цилиндре (т.е. в пробирке – Авт.).
Научно-технические достижения
Примерно 120 лет тому назад В.Н. Ипатьев приступил к реализации опытов по синтезу бутадиена (дивинила), которые были связаны с использованием каталитических реакций, происходящих при высоких температурах и повышенном давлении. Размышляя о причинах небольшого выхода бутадиена, он задумался о роли материала трубки, условиях осуществления реакции и составе продуктов пиролиза. Так была не просто открыта, а осознана роль железа как катализатора. Было выявлено влияние, которое может оказывать материал стенок сосуда на ход реакции (стекло, фарфор, железо). Ипатьев последовательно открыл новые каталитические реакции – разложение алкоголей с образованием альдегида и водорода (1901) и с образованием дивинила (1903). Сделал вывод о том, что не только металл, но и оксид металла должен оказывать каталитический эффект, и открыл глинозём, как катализатор, предложив использовать его в качестве общего способа получения олефинов из алифатических и циклических алкоголей.
 Модель «бомбы Ипатьева»
Модель «бомбы Ипатьева»
В 1903 году, используя свою техническую подготовку военного артиллериста, он создаёт простой в устройстве автоклав, работающий при 500-1000 атм и температуре до 500 градусов. Этот аппарат получил название “бомба Ипатьева”. Им было открыто совместное действие как однотипных, так и разнотипных катализаторов – промотирование.
В 1908 году по особому разрешению Ипатьев был допущен к защите диссертации “Каталитические реакции при высоких температурах и давлениях” в Петербургском университете и успешно её защитил. Так он стал доктором химии. Военный по специальности, профессор и теперь доктор химии!
В годы Первой мировой войны (в начале 1915 г.) при его участии и руководстве была организована Комиссия по заготовке взрывчатых веществ (в апреле 1916 г. преобразованная в Химический комитет) при Главном артиллерийском управлении. Руководителем стал член-корреспондент Академии наук (с 1914 г.), генерал-майор В.Н. Ипатьев. Отдельной строкой следует выделить деятельность Владимира Николаевича по борьбе с удушающими газами, которая проходила в двух направлениях: получение удушающих веществ и создание противогазовых защитных средств. Именно В.Н. Ипатьев стоял во главе всей цепочки по созданию противогазов различных типов (противогаз Горного института, противогаз Куманта-Зелинского, противогаз Химического комитета). Снабжение русской армии в Первой мировой войне взрывчатыми веществами и удушающими средствами было выполнено почти исключительно Химическим комитетом под председательством В.Н. Ипатьева – закономерным стало и присвоение ему звания генерал-лейтенанта.
В конце 1915 года академики Н.С. Курнаков, П.И. Вальден, Б.Б. Голицын предлагают кандидатуру Ипатьева в действительные члены Академии наук, и в 1916 году его утверждают в этом звании.
Деятельность
Химического комитета успешно продолжалась и в период революций 1917
года, но постепенно сокращалась в связи с окончанием военных
действий. В середине 1918 года весь Химический комитет под
председательством В.Н. Ипатьева полностью вошёл в виде отдельной
комиссии в состав Химического отдела ВСНХ (председателем которого был
Л.Я. Карпов). В.И. Ленин называл Ипатьева главой нашей химической
промышленности. В 1927 году Нобелевский лауреат Фриц Габер в своём
поздравлении с 60-летием
Вынужденная эмиграция
Имея обширнейшие связи с мировым научным и техническим сообществом, В.Н. Ипатьев постоянно участвовал в различных международных конференциях, консультировал европейские технические проекты. Большую часть заработанной при этом валюты он вкладывал в советские научные исследования. Тем не менее в СССР начались необратимые процессы по изгнанию и уничтожению интеллектуальной и технической элиты дореволюционной подготовки. Дошла очередь и до “главы химической промышленности”. В 1930 году, спасая свою жизнь, В.Н. Ипатьев вместе с женой выехал из Советской России за границу. Сначала в Германию, а затем в США. В Америке основным местом его работы стал Исследовательский центр фирмы Юниверсал Ойл продактс (Universal Oil Products Company), где он руководил работами по нефтепереработке. В этом центре (г. Риверсайд, штат Иллинойс) фирма оборудовала для него хорошо оснащённую лабораторию высоких давлений. Параллельно основной работе он состоял профессором в Северо-Западном университете близ Чикаго (Northwestern University).

В.Н.
Ипатьев (эмигрант из России) и Г. Пайнс (эмигрант из Польши) в
лаборатории в Чикаго
По утверждению ученика В.Н. Ипатьева академика Г.А. Разуваева, “американцы считают Ипатьева одним из создателей в их стране современной нефтехимии” (Разуваев Григорий Алексеевич (1895-1989), российский и советский химик. В 1934 г. осуждён по “делу славистов” на 10 лет. Академик АН СССР (1966), заслуженный деятель науки РСФСР, основатель Института металлоорганической химии – в настоящее время Институт металлоорганической химии им. Г.А. Разуваева РАН). Американский нефтехимик Фрэнк Уитмор в 1937 году так охарактеризовал научный вклад В.Н. Ипатьева: “В России было три выдающихся химика среди множества великих. Это Ломоносов (крупнейшая в научном мире фигура, основатель университета, носящего его имя), Менделеев (составитель периодической таблицы элементов) и Ипатьев. Причём Ипатьев оказал на развитие химии большее влияние, чем оба его знаменитых соотечественника” [9, p. 78]. В.Н. Ипатьев предложил гетерогенный катализ при высокой температуре и под высоким давлением, чем заложил основы химии ХХ века. Именно его изыскания позволили производить искусственный каучук, пластмассы, моющие средства и многое другое. За 60 лет своей научной деятельности он написал почти 400 статей и десятки книг, среди которых были учебники (в том числе применявшиеся в школьном курсе), запатентовал около 300 открытий, причём в каждом патенте он указывал, что для России эти открытия бесплатны. Свои научные статьи он всегда пересылал на родину.
 Последняя работа В.Н.Ипатьева, переданная им в Советскую Россию из США Она
вышла в свет в 1936 г. Эту и другие его работы можно было
использовать, но ссылаться на них было нельзя
Последняя работа В.Н.Ипатьева, переданная им в Советскую Россию из США Она
вышла в свет в 1936 г. Эту и другие его работы можно было
использовать, но ссылаться на них было нельзя
Главным вкладом В.Н. Ипатьева в науку является использование крекинга для очистки газа и другие открытия, относящиеся к каталитическим реакциям (особенно при высоких давлениях и температурах), переработке нефти и продуктов её перегонки, а также разработка метода промышленного получения высокооктанового бензина (именно это исследование принесло ему славу человека, выигравшего в 1940 году воздушную войну с Германией над Британскими островами). “Никогда за всю историю химии в ней не появлялся более великий человек, чем Ипатьев”, – так сказал о нём Рихард Вильштеттер (Richard Martin Willstӓtter (1872-1942), химик-органик, лауреат Нобелевской премии по химии 1915 г.), с которым Владимир Николаевич стажировался у А. фон Байера.
Сыну В.Н. Ипатьева – Владимиру Владимировичу – пришлось пройти через тяжелейшие нравственные испытания. Он, химик, продолживший дело своего отца, в середине 1936 года защитил докторскую диссертацию. В декабре того же года академика В.Н. Ипатьева всенародно и громкогласно объявляют “дезертиром социалистического труда” [10], лишают гражданства и звания академика (избранного в Академию наук не при советской власти). (Несколько академиков-химиков не явились на процедуру шельмования своего коллеги – Авт.) Требуют, чтобы его сын публично отрёкся от отца. На такой компромисс Владимир Владимирович не пошёл, он лишь выразил огорчение по поводу того, что академик Ипатьев не вернулся в СССР. Это ему дорого обошлось: арест, болезнь, заключение, работа в “шарашке” (химики в условиях начавшейся войны были нужны), ссылка, невозможность жить в Ленинграде и работать в университете после окончания срока наказания – только через 20 лет стало легче.
Его дочь – внучка В.Н. Ипатьева – Варвара Владимировна, названная в честь бабушки, которую она никогда так и не увидела, только в 1991 году по приглашению руководства фирмы Юниверсал Ойл продактс смогла приехать в Чикаго и посетить рабочий кабинет своего дедушки. Она тоже была химиком, кандидатом наук.
Эпилог, но не окончание
 В.Н. Ипатьев с В.Д. Ипатьевой (урожд. Ермаковой)
В.Н. Ипатьев с В.Д. Ипатьевой (урожд. Ермаковой)
Владимир Николаевич умер в 1952 году, через десять дней после его смерти ушла из жизни его верная спутница – жена. Оба они похоронены недалеко от Чикаго. Могила сохранилась. В университете г. Эванстон (Evanston) создан музей, а лаборатория катализа и высоких давлений, которую Ипатьев создал на свои сбережения и в которой работал, носит его имя (Ipatieff High Pressure Laboratory). Здесь все эти годы бережно хранят память о нём.
В Санкт-Петербурге сейчас живут правнук, праправнук и праправнучка академика В.Н. Ипатьева. Химиков среди них нет.
В 1990 году Академия наук возвратила В.Н. Ипатьеву звание академика. С 1994 года (один раз в три года) в России вручается учреждённая РАН премия имени академика Ипатьева за заслуги в области технической химии (в США аналогичная премия существует с 1939 г.). В ноябре 2017 года в Санкт-Петербургском государственном университете (в библиотеке Химического общества) открылась выставка, посвящённая 150-летию со дня рождения В.Н. Ипатьева. Тем не менее нет памятных мемориальных досок ни в военной Михайловской артиллерийской академии, где учился, преподавал и занимался научной и военно-технической деятельностью Владимир Николаевич с 1889 по 1930 гг., ни на здании, где была его академическая квартира и лаборатория будущего института высоких давлений, в которой он жил и работал с 1923 по 1930 гг. (Васильевский остров, 8 линия, д. 17, с мемориальными досками в честь А.М. Бутлерова и Н.Н. Бекетова), ни в Петербургском университете в здании химического института, где читал лекции в начале XX века молодой приват-доцент Ипатьев. Открытой В.Н. Ипатьевым реакции дегидрирования и дегидратации этанола, приводящей к получению бутадиена-1,3, следует присвоить именование “способ Ипатьева-Лебедева”.
Пора снять покров незаслуженного забвения с трудов и жизни величайшего химика России!
Благодарности
Эта статья могла быть подготовлена только при содействии всех ветвей рода Ипатьевых-Черкасовых, поэтому автор искренне благодарит: Андрея Николаевича Черкасова, Николая Андреевича Черкасова, Марию Дмитриевну Данилову (урождённую Гельцер), которые не только предоставили иконографический материал, но подробно познакомили с имеющимися в их распоряжении архивными материалами. Автор также благодарит сотрудников библиотеки Российского химического общества (Библиотека Академии наук России), Российской Национальной библиотеки и музея в Эванстоне, Музея истории Санкт-Петербургского университета.
“Вы, русские, не представляете себе, кого вы потеряли в лице Ипатьева, не понимаете даже, кем был этот человек. Каждый час своей жизни здесь, в США, всю свою научную деятельность он отдал России. Беспредельная любовь к Родине, какой я никогда и ни у кого из эмигрантов не видел, была той почвой, на которой произрастали все выдающиеся результаты исследовательских трудов Ипатьева”, – так обращается к нам ассистент В.Н. Ипатьева профессор Г. Пайнс [9]. Понимаем ли мы, кого забыли?
Нравственные уроки химика В.Н. Ипатьева
Восемьдесят пять прожитых лет – весьма достаточное основание для того, чтобы прислушаться к мнению долгожителя. Можно ли в XXI веке взять что-либо из опыта Владимира Николаевича Ипатьева, тем более, что опыт этот – наше национальное достояние, никак не меньше. Назовём этот опыт нравственными уроками Ипатьева. Вот они:
• Чувство зависти присуще нам всем, все мы имеем этот недостаток, но надо различать два вида этого чувства: я могу завидовать успеху какого-нибудь большого химика, когда он подметил интересное явление в том процессе, который был предметом моего изучения, но в котором я этого явления не заметил. Такая зависть может быть оправдываема, если она не сопровождается порчей отношений между людьми. Но если человек начинает завидовать другому в его успешной работе, когда он сам не хочет или не может отдать свои силы и уменье на продолжительный и упорный научный труд, чтобы выявить свой талант в исследовании новых явлений, то подобная зависть может вызвать только отталкивающее впечатление и установление недоверия между людьми [11, c. 182].
• В научных делах нельзя приписывать себе того, что принадлежит другим [11, c. 200].
• Главная заслуга принадлежит тому, кто сумел после открытия нового явления указать на его важное значение и так его исследовать, чтобы всем стало ясно, какую пользу оно принесёт как для дальнейшего развития науки, так и для пользы людской [3, c. 211].
• Мы, учёные, должны быть очень скромны при оценке наших научных достижений и должны всегда сознавать, что, хотя мы и посвятили всю свою жизнь науке, мы могли внести в её достижения лишь небольшую лепту, так как её задачи безграничны. Кто с любовью вёл научную работу и мог большую часть своей жизни посвятить научным исследованиям, уже тем самым получает величайшее удовольствие, а если он мог передать свои идеи другим для дальнейшей их разработки, то едва ли в какой-либо другой деятельности он нашёл бы большее удовлетворение. Опубликовывая свои исследования, учёный навсегда заносит своё имя на страницы истории науки; он вправе гордиться своей работой, и нельзя упрекать его в том, что только одно честолюбие принуждало его к открытиям в научной области.
Если человек является истинным учёным по призванию, то в тайниках его разума обязательно гнездятся творческие мысли, которые неустанно толкают его в область научных изысканий, и никакие обстоятельства жизни, никакие житейские невзгоды не могут отвратить этого талантливого или гениального творца от реализации его смелых, порою фантастических замыслов [3, c. X–XI].
Литература
Researchers get the first detailed look at how catalysts crank out individual polymer chains
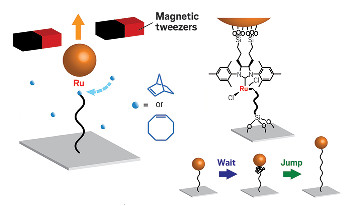
By attaching a polymer chain to a glass slide and to a magnetic particle
bound to the ruthenium catalyst (linkage shown at right) and pulling
the chain out with a pair of magnetic tweezers, researchers are able
to watch the wait and jump dynamics of ring-opening polymerization.
When chemists think about polymerization, they typically envision a wormlike polymer growing smoothly and continuously from a catalyst. But the actual view of how polymer growth unfolds has remained murky because of the limitations of analytical techniques.
Using a pair of magnetic tweezers, optical microscopy, and spectroscopic techniques, Cornell University researchers led by Peng Chen , Geoffrey W. Coates and Fernando A. Escobedo have achieved the first real-time visualization of single polymer chain growth. What they report is startling: Individual polymer chains don’t increase steadily but instead undergo consecutive wait and jump steps.
With the aid of molecular dynamics computer simulations, the researchers attribute this jerky mechanism to formation of polymer tangles—which they call hair balls—that form around the catalyst as thousands of new monomer units are added to the growing chain. The hair balls sporadically unravel after a couple of minutes, and a new hair ball starts to form.
Besides helping researchers better understand catalyst activity, polymerization rates, and bulk polymer properties, the researchers suggest their discovery of the growth spurts may be relevant to how cells produce biopolymers such as proteins, nucleic acids, and polysaccharides (Science 2017, DOI: 10.1126/science.aan6837).
These new findings are “very cool,” says Suzanne A. Blum of the University of California, Irvine. Blum’s group has used fluorescently labeled molecules to study single-molecule dynamics and recently used this approach to watch catalytic activity as labeled monomers randomly got added to a growing polymer chain (Angew. Chem. Int. Ed. 2017, DOI: 10.1002/anie.201708284).
were previously obscured by “ensemble averaging,” Blum explains, in which researchers use techniques such as dynamic light scattering to observe all the molecules in a sample at once, and information about size distributions and other polymer parameters are extracted from the data. Single-molecule approaches avoid limitations of ensemble averaging, Blum says, but these measurement techniques are often a double-edged sword because the resulting data are a challenge to interpret without corroborating spectroscopic methods. By including molecular dynamics simulations, the Cornell team was able to get a clearer picture of conformational changes in the growing polymer.
“The ability to see dynamics in an important reaction like polymerization and to understand them through modeling is an exciting technological advance,” Blum says.
In their single-molecule experiment, the Cornell researchers attached the free end of a polymer chain to a glass surface using a silane linkage and attached the ruthenium catalyst at the growing end of the polymer to a magnetic particle held in place by a pair of magnetic tweezers. By tracking the position of the magnetic particle, the team achieved real-time visualization of a single chain’s growth during ring-opening polymerization.
“This is just a superb piece of science,” says Craig J. Hawker of the University of California, Santa Barbara. Hawker’s group recently reported the one-pot synthesis of block copolymers using five different monomers with widely varying properties (Angew. Chem. Int. Ed. 2017, DOI: 10.1002/anie.201707646). The new polymer-growth monitoring process could help researchers understand how to better control such exotic polymerizations and allow them to tune the macroscopic properties of polymer networks to design new functional materials, he says.
“The Cornell team’s tantalizing view of how polymer chains grow sheds light on many synthetic challenges dating from the earliest days of polymer chemistry,” Hawker adds. “This work will have major implications far beyond single-chain dynamics.”
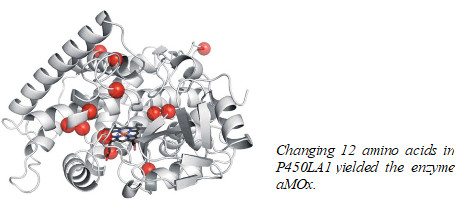
Directed evolution yields enzyme that oxidizes alkenes in a unique way
Oxidizing the end carbon atom of a terminal alkene to form an aldehyde, a process called anti-Markovnikov alkene oxidation, is difficult because the reaction is energetically unfavorable. The preferred process, in terms of energetics, is Markovnikov selectivity—oxidation of the substrate’s next-to-last carbon, forming a ketone instead of an aldehyde.
Chemists have long wanted to find better ways to achieve anti-Markovnikov selectivity in alkene oxidations—for example, to convert the aldehyde products to linear alcohols, which are commonly used in flavorings, perfumes, lubricants, and cosmetics. Catalysts exist that can perform the tricky anti-Markovnikov oxidations, but they are very unproductive and are not enantioselective.
Frances H. Arnold and coworkers at Caltech thought a modified enzyme might succeed where traditional catalysts have struggled. The team now reports using directed evolution, an iterative process of protein mutation and screening for activity, to identify amino acid substitutions that convert an enzyme into a predominantly anti-Markovnikov alkene oxidation catalyst (Science 2017, DOI:10.1126/science.aao1482).
The evolved enzyme, called aMOx, is fairly productive, catalyzing about 3,800 oxidation reactions, or turnovers, before running out of steam, compared to fewer than 10 for earlier anti-Markovnikov oxidation catalysts. In conjunction with other reagents and catalysts, the enzyme could one day convert terminal-alkene substrates to a variety of aldehyde, alcohol, and amine products for commodity and fine chemical, pharmaceutical, and agrochemical use.
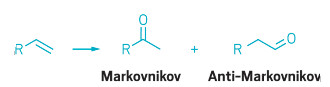
Markovnikov oxidation of terminal alkenes (left) to form ketones (center) is energetically
favored, whereas anti-Markovnikov reactions leading to aldehydes are
more difficult.
The Caltech researchers started with an iron-based cytochrome P450 enzyme called P450LA1 because it catalyzes a related reaction, metal-oxo alkene oxidation, in which a metal-linked oxygen attacks the double bond, typically forming an epoxide. An earlier study by another group had found that P450LA1 also made 19% aldehyde side-product by opening the epoxide. In the new study, postdoc Stephan C. Hammer, now at the University of Stuttgart, found that the enzyme actually made 45% aldehyde directly, without going through an epoxide intermediate.
The researchers then used 10 rounds of directed evolution, with styrene as a substrate, to boost anti-Markovnikov selectivity from 45% to 81%, in aMOx. aMOx works well because it stabilizes high-energy radical and carbocation intermediates created when its iron-oxo group attacks styrene’s terminal alkene, allowing anti-Markovnikov aldehyde formation to predominate.
It took 12 mutations distributed throughout the protein to transform P450LA1 into aMOx. “No one could explain how these substitutions confer this reaction selectivity, much less predict them,” Arnold says. “This illustrates the power of directed evolution to create catalysts that have eluded chemists.”
Key advantages of the evolved enzyme include its enantioselectivity and its turnover number, “which probably still isn’t sufficient for commercial viability but pretty respectable,” says John T. Groves of Princeton University, who specializes in biomimetic catalysis.
There’s currently a push in academia and industry to replace conventional catalytic reactions with enzymatic processes that use aqueous solvents at ambient temperatures and pressures, work readily in flow systems, and don’t require catalyst separations, Groves says. “These days, a tie will go to the biocatalyst for those reasons. The new study is an important step in the right direction.”
Powerful acid family enables cycloaddition of unactivated aldehydes and dienes
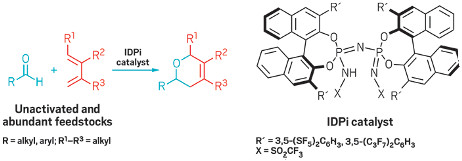
Chiral IDPi catalysts expand the scope of hetero Diels-Alder
reactions
beyond electronically biased partners.
The Diels-Alder reaction is a classic transformation that typically marries a diene (with four π electrons) and an alkene (with two π electrons) to form a six-membered ring—it’s known as a [4+2] cycloaddition. This reaction has been studied exhaustively in organic synthesis and tried in many variations, yet some reacting partner combinations remain out of reach.
Researchers led by Benjamin List at the Max Planck Institute for Kohlenforschung have now closed one of these gaps. The team has reported a method that brings together unactivated and inexpensive dienes and aldehydes for the first time in a hetero Diels-Alder reaction, a variant that trades a carbon atom in the alkene partner for a heteroatom, which is oxygen in the case of the aldehyde (J. Am. Chem. Soc. 2017, DOI: 10.1021/jacs.7b08357).
Diels-Alder reactivity is ruled by the energy gap between the two reacting partners. Chemists need to narrow this gap to drive the reaction forward, generally by modulating the reaction partners’ electronic properties through different substituent groups and/or with catalysts. For hetero Diels-Alder reactions between dienes and aldehydes, success has been limited to electronically engineered reacting partners.
By using highly acidic chiral imidodiphosphorimidate (IDPi) catalysts, List and coworkers were able to access electronically unactivated dienes and aldehydes to form dihydropyrans, which are often found in pharmaceutical and agrochemical settings. The researchers propose that the catalysts create a confined environment that enables high stereocontrol and avoids a number of acid-promoted side reactions.
Efforts to measure the new IDPi catalysts’ acidity are ongoing, but the compounds are estimated to be more acidic than an analogous IDPi compound that is reportedly the strongest chiral acid ever synthesized, according to the researchers.
“I really think this catalyst class is the most significant contribution to come from my lab,” List says. He believes the catalysts hold “enormous potential” for asymmetric synthesis because they are so active and selective.
The new class of strong IDPi acids are “extremely effective” at producing dihydropyrans in high enantiomeric excess, says Varinder Aggarwal, a synthetic chemist at the University of Bristol. Aggarwal points out that these dihydropyran products are valuable in the fragrance industry, adding that List’s lab must be “smelling pretty good.”
Chemical & Engineering News
|
Conference on Advances in Catalysis for Energy and Environment (CASEE 2018) Mumbai, India |
|
4th International Symposium on Chemistry for Energy Conversion and Storage (CHEMENER 2018) Berlin, Germany |
|
2nd Caribbean Conference on Functional Materials (CARIBMAT 2018) Cartagena de Indias, Colombia |
|
Lignofuels 2018 Conference (Advanced Biofuels & Materials) Amsterdam, The Netherland |
|
5th European Biopolymer Summit Dusseldorf, Germany |
conference-europe |
2nd International Conference on Catalysis and Chemical Engineering (CCE 2018) Paris, France |
|
3rd Molecules and Materials for Artificial Photosynthesis Conference Cancun, Mexico |
|
19th Netherlands' Catalysis and Chemistry Conference Noordwijkerhout, The Netherlands |
|
International Conference of Computational Methods in Sciences and Engineering 2018 (ICCMSE 2018) Thessaloniki, Greece |
|
3rd SynGas Convention “Fuels and Chemicals from Synthesis Gas: State of the Art” Cape Town, South Africa |
|
15th International School-Conference on Magnetic Resonance and its Applications (Spinus 2018) St. Petersburg, Russia |
|
27th Biennial Conference of Organic Reactions Catalysis Society San Diego, CA, USA |
|
Designing Nanoparticle Systems for Catalysis Faraday Discussion London, UK |
|
III Всероссийская конференция с международным участием “Исследования и разработки в области химии и технологии функциональных материалов” Апатиты, Россия |
577-konferentsii-2018 |
III Scientific-Technological Symposium “Catalytic Hydroprocessing in Oil Refining” (STS HydroCat-2018) Lyon, France |
|
3rd Green & Sustainable Chemistry Conference Berlin, Germany |
green-and-sustainable-chemistry-conference |
Designing Nanoparticle Systems for Catalysis. Faraday Discussion London, UK |
http://www.rsc.org/events/detail/25362 |
5th International School-Conference on Catalysis for Young Scientists “Catalyst Design: From Molecular to Industrial Level” Moscow, Russia |
|
13th International Symposium on the Synthesis and Applications of Isotopes and Isotopically Labelled Compounds Prague, Czech Republic |
|
10th European Meeting on Solar Chemistry and Photocatalysis: Environmental Applications (SPEA10, 2018) Almeria, Spain |
|
1st International Conference on Reaction Kinetics, Mechanisms and Catalysis Budapest, Hungary |
|
2nd International Symposium on Clean Energy from Ethanol (ISCEE 2018) Krakow, Poland |
|
20th International Conference on Catalysis (ICC 2018) Dubai, UAE |
|
EFCATS School on Catalysis Liblice Castle, Czech Republic |
|
The World Conference on Carbon (Carbon 2018) Madrid, Spain |
|
Post-graduate Summer School on Green Chemistry Venice, Italy |
|
12th International Symposium on Scientific Bases for the Preparation of Heterogeneous Catalysts (PREPA12) Louvain-La-Neuve, Belgium |
|
21th International Symposium on Homogeneous Catalysis (ISHC-21) Amsterdam, The Netherlands |
|
27th IUPAC International Symposium on Photochemistry Dublin, Ireland |
|
4th International Symposium on Catalysis for Clean Energy and Sustainable Chemistry (CCESC 2018) Bilbao, Spain |
|
18th International Symposium on Relations between Homogeneous and Heterogeneous Catalysis (ISHHC 2018) Sydney, Australia |
|
World Congress & Expo on Chemical Engineering & Catalysis (WCECEC-2018) Osaka, Japan |
|
International Symposium on Zeolites and Microporous Crystals (ZMPC 2018) Yokohama, Japan |
|
8th Tokyo Conference on Advanced Catalytic Science and Technology (TOCAT8) Yokohama, Japan |
|
Nordic Symposium on Catalysis Copenhagen, Denmar |
|
7th EuCheMS Chemistry Congress Liverpool, UK |
|
XII Международная конференция молодых ученых по нефтехимии Звенигород, Россия |
|
IV Междисциплинарный симпозиум по медицинской, органической и биологической химии и фармацевтике (МОБИ-ХимФарма2018) Новый Свет, Крым, Россия |
|
15th International Conference on Micro Reaction Technology (IMPET 2018) Karlsruhe, Germany |
|
XXIII International Conference on Chemical Reactors (CHEMREACTOR-23) Ghent, Belgium |
|
6th European Conference on Environmental Applications of Advanced Oxidation Processes (EAAOP-6) Portorose, Slovenia |
|
50th General Assembly & 47th IUPAC World Chemistry Congress Paris, France |
|
14th EuropaCat – European Congress on Catalysis “Catalysis without Borders” Aachen, Germany |
|

